Основания для применения фолиниевой кислоты при нейропсихиатрических заболеваниях
Перевод гипотезы, опубликованной в журнале Biochimie в 2016 году группой Ramaekers et al. (смотрите также новость на связанную тему)
В переведенной статье изложена гипотеза, для доказательств которой нужны более качественные исследования; более взвешенный, классический взгляд на церебральную фолатную недостаточность - в очень хорошем обзоре 2019 года "Cerebral folate deficiency: Analytical tests and differential diagnosis" (Pope et al.) (русский перевод на нашем сайте)
Аннотация
Развитие нейропсихиатрических расстройств происходит под воздействием множества факторов, как генетических, так и экзогенных (наркотики, токсины, психологические травмы). Часть этих заболеваний может сопровождаться системной фолатной недостаточностью – в таких случаях у пациента развивается макроцитарная анемия и отмечаются определенные нейропсихиатрические признаки. У некоторых пациентов, несмотря на нормальные уровни фолата в организме в целом, нарушен транспорт фолата в мозг – это приводит к развитию синдромов, объединяемых под названием «церебральная фолатная недостаточность» (ЦФН). Внешне эти синдромы проявляются как нарушения развития и психические заболевания. Список этих синдромов включает ЦФН с манифестацией в младенческом возрасте, ранний детский аутизм с неврологическими нарушениями либо без них, синдром спастической атаксии, резистентную эпилепсию у детей раннего возраста, резистентную шизофрению у подростков, и резистентную депрессию у взрослых.
В большинстве случае синдром ЦФН развивается из-за аутоиммунной реакции в отношении рецептора фолиевой кислоты альфа (FRa), которая приводит к снижению уровней 5-метилтетрагидрофолата (MTHF) в спинномозговой жидкости (ликворе), в редких случаях – из-за мутаций гена FRa и дефектов митохондриальной ДНК. Возраст, при котором начинается выработка антител блокирующего типа к FRa, определяет клиническую картину синдрома. У пациента с синдромом ЦФН, начавшимся в младенческом возрасте, либо с аутизмом, сопровождающимся неврологическими нарушениями, обычно отмечается повышенный титр антител к FRa и сниженный уровень MTHF в ликворе. Напротив, в случае раннего детского аутизма либо резистентной шизофрении нарушения поведения и другие симптомы могут усиливаться и ослабевать вместе с повышающимся и снижающимся титром антител к FRa, сопровождающимся колебаниями уровней фолата, тетрагидробиоптерина (BH4) и нейромедиаторных метаболитов в спинномозговой жидкости между низкими и нормальными уровнями. Мы предлагаем к рассмотрению гипотетическую модель, объясняющую патогенез шизофрении.
Вооружившись результатами, полученными в ходе клинических исследований, исследований генома, анализа спинномозговой жидкости и МРТ-данных, мы рассмотрим нейрохимические отклонения, отмечаемые при данных расстройствах, метаболические и регуляторные каскады, процессы синтеза и катаболизма нейромедиаторов, а также воздействие окислительного стресса на патогенез данных расстройств.
Мы предлагаем к изучению диагностический алгоритм и терапевтические схемы, предполагающие назначение кортикостероидов, больших доз фолиниевой кислоты и безмолочной диеты. Было доказано, что эти меры способствуют нормализации уровня фолата в мозге и снижению титра аутоантител к FR у лиц с аутоиммунной реакцией.
1. Введение
У взрослых пациентов с дефицитом фолиевой кислоты или витамина B12 может развиться мегалобластная анемия, но это происходит не всегда. Развитие нейропсихиатрических симптомов может случиться как до, так и после развития мегалобластной анемии. У взрослых пациентов с мегалобластной анемией неврологические симптомы дефицита фолиевой кислоты и дефицита витамина B12 разительно схожи и включают периферическую нейропатию, патологию спинного мозга, известную под названием «подострая комбинированная дегенерация», и атрофию зрительного нерва. По результатам нейрофизиологических исследований периферических нервов у пациентов с дефицитом фолиевой кислоты в основном выявляется сенсорная аксональная нейропатия.[4,5] Психиатрические симптомы, выявляемые у пациентов с дефицитом фолиевой кислоты, обычно включают снижение когнитивных функций и отдельные когнитивные нарушения, симптомы депрессии и шизофрении, в то время как при дефиците витамина B12 в основном отмечается снижение когнитивных функций. Нейропсихологическое тестирование пациентов с дефицитом фолиевой кислоты выявляет нарушения внимания, эпизодической и зрительно-пространственной памяти и абстрактного мышления.[6-11] Клиническая картина при системном дефиците фолиевой кислоты зависит от возраста конкретного пациента на момент начала заболевания, от длительности и выраженности дефицита, от наличия предрасполагающих генетических факторов, в том числе полиморфизмов генов, кодирующих фолат- и B12-зависимые ферменты и генов, оказывающих влияние на соответствующие метаболические цепочки [1].
Врождённая мальабсорбция фолиевой кислоты – аутосомно-рецессивное заболевание, при котором мутации, нарушающие работу протон-сопряженного транспортера фолатов (proton-coupled folate transporter, PCFT), приводят к развитию дефицита фолиевой кислоты с нарушенной абсорбцией фолатов в кишечнике и нарушением переноса фолатов в центральную нервную систему (ЦНС). Наблюдаемые при этом симптомы включают в себя вялое сосание и недостаточную прибавку в весе, развитие мегалобластной анемии, часто сопровождающейся лейкопенией и/или тромбоцитопенией, развитие диареи и/или воспаления слизистой оболочки полости рта, развитие гипоиммуноглобулинемии и других нарушений иммунной системы, приводящих к заражению нетипичными бактериями. Неврологические проявления включают в себя задержку в развитии, гипотонию и атаксию, умственную отсталость, дискинезии (атетоз). Часто наблюдаются эпилептические приступы, также отмечается периферическая нейропатия и кальцификация базальных ганглиев[12,13]. Схожие неврологические симптомы наблюдаются при так называемой «церебральной фолатной недостаточности с началом в младенческом возрасте» - они начитают проявляться в возрасте от 4 до 6 месяцев. Вслед за открытием младенческой формы синдрома ЦФН нам удалось обнаружить разновидности синдрома с более поздним дебютом, при которых также снижено содержание MTHF в ликворе. В связи с этим было избрано следующее определение ЦФН: "любое нейропсихиатрическое состояние, при котором наблюдается снижение уровней MTHF в ликворе при сохранном фолатном гомеостазе за пределами центральной нервной системы." Возраст, в котором в мозге развивается недостаточность фолата, в сочетании с пока еще неустановленными генетическими факторами, влияет на то, какими будут первые проявления синдрома, его симптомы, и предопределяет различия между различными подвидами синдрома ЦФН [16] (рис.1).
Мембранносвязанные FRа связывают MTHF для перемещения фолата внутрь эпителиальных клеток сосудистого сплетения, после чего осуществляется транспорт фолата к другому краю клетки и перенос фолата в спинномозговую жидкость. После этого фолат опять проходит сквозь клетки – на этот раз сквозь клетки эпендимы – и попадает в нейрональное межклеточное пространство [16] (рис. 2). Протон-сопряженный транспортер фолатов, вероятно, участвует в процессе высвобождения фолата, связанного с FRa в подкисленных везикулах. Проведенные ранее исследования показали, что основной причиной развития ЦФН у младенцев является аутоиммунная реакция в отношении FRa. Циркулирующие аутоантитела к FR связываются с FRa, прикрепленными к мембранам эпителиальных клеток сосудистого сплетения, и попадают внутрь клеток вследствие эндоцитоза. FR-аутоантитела блокирующего типа способны блокировать фолат-связывающий карман рецептора, в то время как FR-антитела связывающего типа прикрепляются к другим белковым участкам фолатного рецептора – это запускает каскад реакций системы комплемента, вызывает активацию цитокинов, и, в итоге, приводит к разрушению комплекса антиген-антитело [19]. Генетические отклонения, затрагивающие ген FR, встречаются редко. В результате таких отклонений нарушается форма рецептора, вследствие чего он либо не закрепляется на мембране должным образом, либо не выполняет свои функции. [20, 21] Также сообщалось случаях развития ЦФН в у пациентов с митохондриальными цитопатиями [22-24]. Каждый третий пациент с младенческой формой синдрома ЦФН, вызванной антителами к FR, удовлетворяет диагностическим критериям низкофункционального аутизма. У целого ряда девочек в этой группе пациентов наблюдаются все клинические признаки синдрома Ретта, несмотря на нормальные результаты генетического анализа. В исследованиях было обнаружено, что вне зависимости от генотипа по гену MECP2, у 42% пациенток европейского происхождения с синдромом Ретта (n=33) отмечается снижение уровней MTHF в ликворе, при этом у 24% пациенток обнаруживаются блокирующие антитела к FRa [25, 26].
После обнаружения ЦФН, вызванной аутоантителами к FR, у мальчика, страдающего аутизмом, и его неполнородной сестры, страдающей резистентной детской шизофренией, мы инициировали программу скрининга, в ходе которой проводился анализ ликвора и выявление аутоиммунных реакций в отношении фолатного рецептора в выборке, включавшей пациентов с ранним детским аутизмом без неврологических нарушений и пациентов с психозом, резистентной шизофренией, шизоаффективными расстройствами и большим депрессивным расстройством [27, 28]. В настоящем обзоре мы представим краткое описание различных синдромов ЦФН, а затем сосредоточимся на психических расстройствах, при которых в центральной и периферической нервных системах может наблюдаться фолатная недостаточность.
2. Синдромы церебральной фолатной недостаточности
Следует провести разделительную черту между сочетанием факторов внешней среды и генетических факторов, приводящим ко всему множеству существующих нейропсихиатрических расстройств, и подгруппой состояний, вызываемых системной недостаточностью фолата и витамина B12 – такие состояния можно диагностировать по наличию макролитической анемии и нескольких нейропсихиатрических фенотипов [1]. В период беременности, эмбриону и плоду жизненно необходимо получать достаточные количества фолата и витамина B12 для нормального развития и предотвращения дефектов нервной трубки (ДНТ) и, возможно, некоторых других врожденных пороков развития [29-31]. Признано, что фолатная недостаточность во время беременности повышает риск развития дефектов нервной трубки и расстройств аутистического спектра у ребенка [32, 33]. Однако если у беременной женщины в сыворотке крови присутствуют антитела к FRa, то даже в случае нормального фолатного статуса может нарушиться перенос фолата через плаценту, повышая риск развития тех же осложнений беременности [34, 35].
Антитела к FRa способны нарушить перенос фолата в мозг и после родов [17, 18]. У пациентов с младенческой формой ЦФН, которая была описана первой, синдром проявляется в 4-месячном возрасте такими симптомами, как 1) выраженное беспокойство, раздражительность и бессонница; 2) замедление роста головы; 3) психомоторная заторможенность и задержка либо регресс развития; 4) гипотония и атаксия; 5) признаки поражения пирамидной системы, отмечаемые в дистальных отделах нижних конечностей; 6) дискинезии (хореоатетоз, баллизм); и 7) эпилепсия. При сравнении со здоровыми испытуемыми, составляющими контрольную группу, у пациентов обнаруживаются нормальные уровни эритроцитарного фолата, витамина B12 и гомоцистеина, но отмечается значительное снижение уровней MTHF в ликворе [14,15] (см. табл. 1).
В зависимости от возраста, в котором организм пациента начинает генерировать антитела к FRa, наблюдаются разные клинические фенотипы синдрома: это может быть ЦФН с дебютом в младенческом возрасте [14,15], ранний детский аутизм с неврологическими нарушениями [36], синдром Ретта, иные расстройства аутистического спектра [25,26,37,38], синдром спастической атаксии [39], резистентная эпилепсия в раннем детском возрасте [40-42], резистентная шизофрения в подростковом возрасте [27,28], и, наконец, резистентное большое депрессивное расстройство во взрослом возрасте [ему посвящен один из разделов ниже по тексту] (рис. 1). То, что у пациентов с аутизмом, в организме которых обнаруживаются высокие титры блокирующих антител к FRa, также отмечается повышение ТТГ [28], нельзя объяснить воздействием антител на щитовидную железу, поскольку клетки щитовидной железы как в детском, так и во взрослом возрасте экспрессируют крайне небольшое количество FRa либо совсем не экспрессируют данный рецептор. В то же время у большинства детей отсутствуют клинические признаки гипотиреоза: уровни гормонов T3 и T4 в сыворотке у таких пациентов не выходят за рамки нормального диапазона, так что повышение уровней ТТГ остается необъясненным.
Для пациентов с младенческой формой синдрома ЦФН, а также для тех, у кого синдром проявляется в виде аутизма с неврологическими нарушениями, характерно наличие следующих признаков: ранний дебют заболевания, устойчивый характер неврологических нарушений, повышенные титры антител к FRa, сниженный уровень MTHF в ликворе. Напротив, у пациентов с расстройствами аутистического спектра без неврологических нарушений, с резистентной шизофренией, шизоаффективным расстройством либо депрессией признаки и симптомы возникают в более позднем возрасте и могут значительно изменяться со временем. Предположительная причина этого – в том, что титры антител к FR колеблются, и параллельно этому содержание MTHF, BH4 и нейромедиаторных метаболитов также колеблется между сниженным и нормальным уровнем [16,27].
2.1. Эпидемиологические данные
С возрастом распространенность блокирующих антител к FRa повышается из-за процессов старения иммунной системы (immunosenescence): в то время как в возрастной группе <16 лет доля лиц с антителами составляет 2%, в популяциях здоровых женщин взрослого возраста в Испании, Ирландии и США доля лиц с антителами составляет 4%-7%, 9%-13% и 10%-15% соответственно[16]. С другой стороны, наличие низкого титра антител у небольшой части здоровой взрослой популяции, судя по всему, не связано с развитием заболеваний. В детском возрасте наблюдается обратная корреляция между титрами блокирующих аутоантител к FRa и уровнями MTHF в ликворе, поскольку при блокировке аутоантителами более 0.25 пмоль FRa/мл сыворотки уровень MTHF в ликворе начинает падать.
Первая описанная форма синдрома ЦФН, 90% случаев которой связывают с воздействием аутоантител к FRa и дебют которой происходит в младенческом возрасте, - орфанное заболевание с оценочной распространенностью от 1/40000 до 1/60000 детей, одинаковой заболеваемостью среди мальчиков и девочек, одинаковой распространенностью среди всех этнических групп. Поскольку низкофункциональный аутизм отмечается у трети пациентов с синдромом ЦФН младенческого типа, фенотип "аутизм с низким IQ и неврологическими нарушениями", при котором почти всегда обнаруживается высокая концентрация антител к FRa, считается очень редкой находкой в общей популяции пациентов с расстройствами аутистического спектра. Неизвестно, насколько часто встречается разновидность синдрома ЦФН, вызываемая аутосомно-рецессивными дефектами гена FRa, но, вероятно, ее распространенность еще ниже.
Распространенность расстройств аутистического спектра оценивается в 1.5%, она варьирует от 0.6% до 2.2% в разных областях США. Оценки распространенности этих расстройств в Европе колеблются от 1% до 2%.
Примерно каждый третий ребенок с расстройством аутистического спектра страдает низкофункциональным аутизмом с уровнем IQ ≤ 70. Наши исследования показали, что в данной популяции с низкофункциональным аутизмом у 55-59% пациентов обнаруживаются аутоантитела к FRa блокирующего типа. Наши результаты были подтверждены группой Frye et al., обнаружившей аутоантитела к FRa у 60% детей с расстройствами аутистического спектра (38).
Распространенность шизофрении среди лиц старше 18 лет оценивается в 1%. После первого года с момента постановки диагноза около 20% пациентов переживает рецидив, несмотря на адекватную терапию с использованием антипсихотиков. Поскольку крайне маловероятно то, что те 80% пациентов с шизофренией, чьи симптомы поддаются терапии антипсихотиками, страдают от ЦФН, вызванной аутоантителами к FRa, мы проводили наши исследования только на пациентах, резистентных к терапии. Мы обнаружили наличие блокирующих аутоантител к FRa у 85% пациентов с резистентной шизофренией. Таким образом, согласно нашим оценкам, распространенность резистентной шизофрении, связанной с наличием аутоантител к FRa, в общей популяции составляет 0.17%. Распространенность большого депрессивного расстройства оценивается приблизительно в 6.7%. Мы проводили исследования только среди пациентов, страдающих большим депрессивным расстройством, среди которых мы обнаружили наличие низких положительных титров аутоантител к FRa у 56%.
2.2 Биохимические отклонения
После всасывания фолатов в кишечнике, большая часть фолиевой кислоты и других фолатов преобразуется в MTHF в энтероцитах подвздошной кишки, и лишь затем покидает эти клетки и попадает в кровоток [43,44]. В связи с этим основная форма фолата, присутствующая в крови – MTHF, и эта же молекула является той формой фолата, которая проникает сквозь гематоэнцефалический барьер и плацентарные барьеры. Некоторые полиморфизмы гена 5,10-метилентетрагидрофолатредуктазы (MTHFR), такие как гомозиготная мутация C677T, распространенность которой в европеоидных популяциях достигает 15%, снижают активность фермента MTHFR и, как следствие, концентрацию MTHF, что ограничивает поступление фолата в различные органы и в мозг. Исследования носителей гомозиготной мутации 677TT гена MTHFR подтвердили снижение уровней фолата в эритроцитах и в спинномозговой жидкости пациентов. Последующие исследования продемонстрировали также накопление формилированных форм тетрагидрофолата и сниженное содержание MTHF в эритроцитах. У пациентов с тяжелой врожденной формой недостаточности MTHFR в спинномозговой жидкости были отмечены низкие уровни MTHF, биоптерина и неоптерина, а также сниженные уровни метаболитов моноаминовых нейромедиаторов [46]. На рисунке 3 показаны фолат-зависимые метаболические пути и метаболиты.
При церебральной фолатной недостаточности нарушается работа множества метаболических цепочек [1,14-16]. Сниженная ферментная активность MTHF-B12-зависимой метионинсинтазы приводит к накоплению гомоцистеина, оказывающего нейротоксическое воздействие [47], и к истощению S-аденозилметионина (SAM), в результате чего нарушается более 100 SAM-зависимых реакций переноса одноуглеродных фрагментов, в том числе реакций, связанных с синтезом мелатонина, катаболизмом дофамина и эпигенетической регуляцией активности генов [27, 48-50]. Поскольку восстановленные метаболиты фолата участвуют в de novo-синтезе пурина и тимидина, при церебральной фолатной недостаточности нарушается процесс репликации ДНК, транскрипции РНК и синтеза гуанозинтрифосфата, являющегося субстратом для ГТФ-циклогидролазы 1 – фермента, участвующего в синтезе тетрагидробиоптерина (BH4). BH4 является кофактором триптофангидроксилазы, тирозингидроксилазы и NO-синтазы (NOS) – скорость-определяющих ферментов, участвующих в синтезе серотонина, дофамина и оксида азота (NO) соответственно. Анализ спинномозговой жидкости, взятой у пациентов с шизофренией и колеблющимися уровнями аутоантител к FRa в ходе проведенного нами недавно исследования, подтвердил наличие положительной корреляции между уровнями MTHF и BH4. Таким образом, профиль состава ликвора при ЦФН обычно характеризуется сниженными уровнями не только MTHF, но также и BH4, и моноаминовых метаболитов [27].
У множества пациентов повышенная активность прооксидантных факторов и/или сбой антиоксидантных механизмов провоцируют процессы окислительного стресса в ЦНС и за её пределами, что вызывает колебания уровней восстановленных фолатов и приводит к повреждению мембран, содержащих транспортеры фолата. В результате этого замедляются процессы захвата фолата и переноса его через барьеры [51,52]. Таким образом, недостаток фолата в нейронах во время окислительного стресса может косвенным образом нарушать синтез BH4 и оборот нейромедиаторов.
Вдобавок к этому гомоцистеин вызывает целый ряд нейротоксических явлений, в частности, стресс эндоплазматического ретикулума и окислительный стресс, а также приводит к эксайтотоксичности, являясь атипичным стимулятором NMDAr-содержащих нейронов [47,53,54]. Данные нейроны в результате чрезмерной стимуляции NMDA-рецепторов могут запускать программы апоптоза [55-60].
Поскольку работа взаимодействующих друг с другом метаболических циклов, таких как цикл трансметилирования, цикл реметилирования фолата и цикл транс-сульфирования, а также метаболических цепочек, связанных с синтезом нейромедиаторов, зависит, помимо прочего, от наличия адекватных запасов железа, цинка, магния, меди и таких кофакторов, таких как витамины B12, B2, B6 и аминокислоты, истощение данных важных элементов может повлиять как на метаболические превращения, в которых они непосредственно принимают участие, так и на другие метаболические пути, связанные с этими процессами опосредованно [61-67]. Сбой механизмов антиоксидантной защиты либо усиление прооксидантных факторов в таких условиях усугубит окислительный стресс и приведет к еще более тяжелым нарушениям метаболического гомеостаза [68-69].
При МРТ-исследовании мозга пациентов с мутациями гена фолатного рецептора либо повышенным титром аутоантител к FRa и сниженным уровнем фолата в ликворе обнаруживается выраженная гипомиелинизация, а МР-спектроскопическое исследование демонстрирует сниженное содержание холина и инозитола в белом веществе [15,20]. Считается, что при фолатной недостаточности фосфатидилхолин – важный компонент мембран глиальных клеток – покидает мембраны, преобразуется в холин и окисляется до триметилглицина (бетаина), являющегося субстратом фермента бетаин-гомоцистеин-метилтрансферазы. Активность данного фермента представляет собой альтернативный путь реметилирования, превращающий гомоцистеин в метионин [70]. Таким образом, развитие синдромов ЦФН приводит к вторичному отбору холина из нейрональных мембран для обеспечения альтернативного пути реметилирования с участием бетаин-гомоцистеин-метилтрансферазы, что в итоге приводит к истощению запасов холина в нервной системе.
Недавно были получены свидетельства того, что FRa не только играет роль в FRa-опосредованном эндоцитозе фолата – было отмечено, что часть фолатных рецепторов покидает эпителий сосудистого сплетения в экзосомах, преодолевает таким образом эпендиму, после чего экзосомы с FRa захватываются нейронами. Последовавшие за этим исследования выявили наличие фолатных рецепторов вблизи ядер клеток, вызвав предположение, что данный рецептор может служить фактором транскрипции, активирующим определенные группы нейрональных генов.
3. Вызванные аутоантителами к FRa аутизм и шизофрения у членов одной семьи
Шизофрения, ассоциированная с наличием аутоантител к FRa и сниженными уровнями MTHF в ликворе, была впервые описана у женщины, рождение и развитие которой протекало без отклонений. У пациентки была диагностирована резистентная к терапии параноидная форма шизофрении. До этого у младшего неполнородного брата пациентки была выявлена церебральная фолатная недостаточность с дебютом в младенчестве и с признаками аутизма, вызванная аутоиммунной реакцией в отношении фолатного рецептора альфа (генеалогическая информация и результаты анализа спинномозговой жидкости представлены на рис. 4).
Начиная с 17 лет у описываемого первоначального пациента отмечались позитивные симптомы – слуховые галлюцинации, паранойя, а также негативные симптомы и когнитивные отклонения. Симптоматика сохранялась несмотря на назначение антипсихотиков и психотерапии. Дополнительные обследования (сканирование мозга, ЭЭГ, лабораторные анализы) не выявили никаких отклонений.
Несмотря на нормальные уровни фолата, B12 и гомоцистеина в крови, уровень фолата в спинномозговой жидкости был ниже нормы, а уровень птеринов – на нижней границе нормы. В сыворотке пациентки были обнаружены аутоантитела к FRa блокирующего типа. Назначение больших доз фолиниевой кислоты и безмолочной диеты привело к драматическому улучшению состояния женщины, у которой исчезли как негативные, так и позитивные симптомы. Улучшение сохранялось даже после отмены всех препаратов. Через восемь лет состояние пациентки оставалось стабильным, и она училась на социального работника.
Полученные данные побудили нас провести скрининг на наличие ЦФН и аутоантител к FRa среди пациентов, страдающих резистентной шизофренией и низкофункциональным аутизмом с неврологическими отклонениями либо без них.
4. Низкофункциональный аутизм с неврологическими отклонениями либо без них
Мы осуществили исследование в группе, состоящей из 25 пациентов (средний возраст: 5.6, диапазон: 2-13.9 лет), страдавших низкофункциональным аутизмом с наличием как минимум одного неврологического отклонения – картина, типичная для ЦФН младенческого типа [36]. При сравнении испытуемых с контрольной группой не было отмечено отклонений уровней фолата в сыворотке и в эритроцитах, а также уровней витамина B12 и гомоцистеина. У каждого из 25 пациентов была значительно снижена средняя концентрация MTHF в спинномозговой жидкости в сравнении с членами контрольной группы (среднее ± СКО: 22.9 ±17 нмоль/л в сравнении со средним уровнем ± СКО, равным 82 ± 31.3 нмоль/л среди членов контрольной группы). У 24 из 25 пациентов с низким уровнем MTHF в ликворе были обнаружены блокирующие аутоантитела к FRa - средний уровень составил 1.09 пмоль заблокированных FRa/мл сыворотки (диапазон: 0 - 4.19) (см. таблицу 1).
Последующее исследование было проведено на 59 пациентах той же возрастной группы, страдавших низкофункциональным аутизмом без неврологических отклонений. При сравнении с пациентами с аутизмом, сопровождающимся неврологическими отклонениями и с контрольными испытуемыми, у данных 59 пациентов уровни фолата в сыворотке оказались немного ниже. Уровни MTHF в ликворе также оказались умеренно снижены, в отличие от групп пациентов с аутизмом и неврологическими отклонениями и пациентов с младенческой формой ЦФН, у которых были отмечены очень низкие уровни MTHF в ликворе (таблица 1).
Менее выраженное снижение уровней MTHF в ликворе у пациентов с аутизмом без неврологических отклонений может быть связано с тем, что у данных пациентов титры аутоантител к FRa повышены умеренно, в отличие от пациентов с аутизмом и неврологическими отклонениями и пациентов с младенческой формой синдрома ЦФН, у которых повышенные титры антител провоцируют более сильное снижение уровней MTHF в спинномозговой жидкости. Таким образом, подтверждается отмеченная нами ранее обратная корреляция между уровнями аутоантител к FRa в сыворотке и уровнями MTHF в ликворе [72]. В то время как блокирующие антитела к FRa были обнаружены почти у всех пациентов с аутизмом и неврологическими отклонениями, они были обнаружены лишь у 35 из 59 пациентов с аутизмом (59%).
В группе, состоящей из 110 детей более юного возраста (средний возраст ± СКО: 4.8 ± 3.3 года),
получивших диагноз низкофункционального аутизма, была оценена распространенность блокирующих аутоантител к FRa в сыворотке самих детей и их родителей. Было осуществлено сравнение результатов с результатами контрольной группы, состоящей из 30 семей с детьми с задержками развития без черт аутизма. Данное исследование было продолжением проведенного ранее предварительного исследования [34]. У 6 пациентов с аутизмом (5%) были обнаружены генетические отклонения. Согласно полученным результатам, блокирующие аутоантитела к FRa были обнаружены у 55% исследованных детей. Блокирующие аутоантитела к FRa были обнаружены у 25% матерей и 26% отцов детей с аутизмом, в то время как в 30 контрольных семьях аутоантитела к FRa были найдены лишь у 1 ребенка и его родителей. Поскольку в некоторых семьях аутоантитела к FRa обнаруживались у одного либо обоих родителей, но не у ребенка с аутизмом, в итоге полученные нами данные показывают, что в 71% из 110 семей блокирующие аутоантитела к FRa присутствовали у ребенка и/или его родителей, а в 29% семей аутоантитела к FRa отсутствовали как у ребенка, так и у обоих родителей. В 110 семьях наблюдались разнообразные комбинации наличия и отсутствия антител к FRa у ребенка с аутизмом и его родителей (рис. 5). Во многих семьях аутоантитела к FRa были обнаружены только у ребенка/детей с аутизмом, но не были найдены ни у одного из родителей. Однако в одной семье с неродственными родителями турецкого происхождения здоровая мать оказалась носителем блокирующих аутоантител к FRa. Она родила трех дочерей, у каждой из которых были обнаружены аутоантитела к FRa. Девочки страдали аутизмом со сниженным интеллектом, а младшая из них также страдала резистентной формой эпилепсии и демонстрировала некоторые признаки синдрома Ретта. В другой семье носителем аутоантител к FRa оказался здоровый отец. Антитела были обнаружены у его сыновей – дизиготных близнецов, в то время как у мамы мальчиков анализ не выявил наличия антител.
У 16% из 110 семей аутоантитела были обнаружены в сыворотке у одного из родителей (6% отцов и 8% матерей) либо обоих родителей (2%), но не были найдены у ребенка, страдавшего аутизмом. В одной тунисской семье аутоантитела к FRa были обнаружены у обоих родителей, но при этом у их детей – монозиготных близнецов, страдавших тяжелой формой аутизма – анализ не выявил антител. Аутоантитела к FRa в организме матери во время беременности могут затруднить трансплацентарный перенос фолата в организм развивающегося плода, повышая риск развития аутизма у ребёнка. Наличие аутоантител к FRа в сыворотке крови здорового отца может также быть ассоциировано с повышенным риском развития аутизма у ребенка, даже если аутоантитела к FRa отсутствуют в организме матери и ребенка. Хотя постулированный эпигенетический механизм, позволяющий объяснить такое сочетание, еще не был исследован, известно, что аутоантитела к FRa у мужчин проникают в ткани, окружающие мужские яички, что может привести к блокированию переноса фолата в формирующиеся сперматозоиды. Нарушенный метаболизм фолата может привести к снижению синтеза пурина и тимидина, что может спровоцировать пока еще неисследованные эпигенетические отклонения [73, 74].
5. Резистентная шизофрения
После обнаружения ЦФН, вызванной аутоантителами к FRa у одного пациента с резистентной параноидной шизофренией, мы провели скрининг с целью обнаружения блокирующих аутоантител к FRa у 20 пациентов с резистентной шизофренией, проявляющейся позитивными и негативными симптомами и не поддающейся лечению антипсихотическими препаратами и психотерапией. Для исключения наличия у пациентов состояний, маскирующихся под шизофрению, было проведено МРТ-сканирование, анализ ЭЭГ и расширенная серия лабораторных анализов. У всех пациентов были отмечены нормальные уровни гомоцистеина, фолата и витамина B12 в плазме крови. Каждую неделю у пациентов брали образцы сыворотки для анализа на содержание аутоантител блокирующего типа к FRa. Аутоантитела к FRa в сыворотке были обнаружены у 17 из 20 пациентов (85%), в то время как в контрольной группе аутоантитела присутствовали лишь у 1 из 30 человек (3.3%). Титры аутоантител к FRa у пациентов варьировали со временем – снижаясь до негативных либо повышаясь до высокого уровня. Такие колебания должны были повлиять на доставку фолата в спинномозговую жидкость, в соответствии с обнаруженной нами ранее обратной корреляции между уровнями аутоантител к FRa в сыворотке и уровнями MTHF в ликворе. Флуктуации титров аутоантител к FRa также сопровождались флуктуациями результатов анализов спинномозговой жидкости среди 15 пациентов: у 8 из них были обнаружены низкие уровни фолата в ликворе, а у 7 – нормальные. Общий средний уровень MTHF в ликворе был снижен (среднее±СКО: 43.73±25.5 нмоль/л; диапазон: 12-83) в сравнении с контрольным уровнем, определенным ранее (t-критерий: 4.48; p<0.0001). Была отмечена позитивная линейная корреляция уровней MTHF и биоптерина в сыворотке крови. Уровни метаболитов дофамина и серотонина в ликворе были либо ниже нормы, либо на нижней границе нормы (рис. 6). На основании этого мы считаем, что пациентам с резистентной шизофренией показано проведение серии анализов на содержание аутоантител к FRa в сыворотке крови. Предполагается, что уровень аутоантител к FRa и его флуктуации влияют на негативные и позитивные клинические симптомы посредством воздействия на доставку фолата в мозг. Повышение либо снижение промежуточных продуктов метаболизма фолатов в мозге связано с ходом метаболических процессов, сказывающихся на уровнях гомоцистеина, на синтезе тетрагидробиоптерина и нейромедиаторов [27,72].
При благоприятном ответе на терапию фолиниевой кислотой было отмечено не только исчезновение позитивной симптоматики, но и значительное ослабление негативных симптомов. Мы пока еще не знаем, насколько большой вклад в развитие шизофрении вносят обнаруживаемые у пациентов аутоантитела к FRa. Если оставить в стороне собственно генетические факторы предрасположенности, дифференциальная диагностика шизофрении предполагает рассмотрение других возможных нейроиммунных причин заболевания, например, антител к N-метил-D-аспартатному (NMDA) рецептору и к компонентам калиевых каналов [75-77].
Предрасположенность к шизофрении в основном обуславливается генами, что подтверждается семейными анамнезами, но также и внешними факторами, такими как осложнения при родах, социальная изоляция, миграция, жизнь в урбанизированной среде и применение в раннем возрасте таких наркотических средств, как леводопа, кокаин, амфетамины и каннабис [78, 79]. Антипсихотические препараты блокируют D2-рецепторы, улучшая состояние пациентов с шизофренией и подтверждая тем самым раннюю этиологическую гипотезу о гиперактивации дофаминергической нейротрансмиссии либо чрезмерной экспрессии дофаминовых D2-рецепторов в мозге пациентов. Функции генов риска заболевания связаны с активностью факторов роста, участвующих в росте и развитии нервных волокон, а белки, кодируемые генами, наиболее связанными с заболеванием, участвуют в дофамергической и глутаматергической сигнальной активности. Понимание заболевания не ограничивается классической дофаминовой гипотезой: в недавнее время интерес исследователей привлекла гипотеза гипофункции глутаматного NMDA-рецептора [82-83]. Гипотеза, предполагающая наличие дефектов глутаматергической нейротрансмиссии либо дефектов сигнальной функции NMDA-рецепторов, была создана для объяснения того факта, что применение неконкурентных антагонистов NMDA-рецептора кетамина и MK801 вызывает шизофреноподобные симптомы, в то время как агонисты глицинового сайта NMDAR-комплекса, согласно сообщениям, смягчают негативные симптомы шизофрении. В последующих исследованиях были получены дополнительные свидетельства в поддержку гипотезы о гипофункции NMDA-рецептора при шизофрении: у мышей, нокаутных по NMDA-рецептору, были отмечены поведенческие отклонения, ассоциируемые с этим заболеванием [84].
Исследования с использованием методов сканирования мозга, длившиеся 5-10 лет, показали корреляцию прогрессирования когнитивных нарушений с увеличением желудочков мозга и потерей серого вещества. В детальных когортных исследованиях пациентов с шизофренией обнаружился специфический изменяющийся со временем характер потери серого вещества [85]. Потеря серого вещества начинается с теменных областей мозга, отвечающих за зрительно-пространственные функции и ассоциативное мышление. В течение 5 лет область потери разрастается в антериорном направлении в височные доли, в сенсорно-двигательную кору, дорсолатеральную префронтальную кору и фронтальные глазодвигательные поля. Согласно подтвержденным данным, вентромедиальная префронтальная кора и верхняя височная извилина являются последними по очереди областями мозга, в которых происходит потеря серого вещества у взрослых пациентов. Сообщалось об обнаружении аутоантител к FRa у подростков с острым психозом либо шизоаффективным расстройством, однако ввиду ограниченности этих данных требуется осуществление крупных исследований.
5.1 Гипотетическая модель патогенеза шизофрении
В мозге здорового человека нисходящие аксональные ветви глутаматергических нейронов коры стимулируют ингибиторные ГАМК-секретирующие интернейроны в стволе головного мозга (вентральная область покрышки) и тем самым опосредуют тоническое ингибирование мезолимбических дофаминергических нейронов. ГАМКергическое тоническое ингибирование данных мезолимбических дофаминовых нейронов в стволе головного мозга сдерживает дофаминергическую гиперстимуляцию их проекционных зон в стриатуме и лимбической системе. В отличие от клеток, ингибирующих мезолимбические дофаминовые нейроны, кортико-стволовые глутаматергические нейроны соединяются непосредственно с мезокортикальными дофаминовыми нейронами ствола мозга, обеспечивая тоническое возбуждение данных мезокортикальных дофаминовых нейронов, аксональные терминалы которых посылают импульсы в дорсолатеральную и вентромедиальную префронтальную кору [82].
Согласно самой ранней версии дофаминовой гипотезы, позитивные симптомы шизофрении отражают чрезмерную активность мезолимбических дофаминовых нейронов вентральной покрышки, которая вызывает гиперстимуляцию проекционных областей данных нейронов в стриатуме и лимбической системе [86-89]. В свою очередь, негативные симптомы шизофрении являются отражением недостаточной активности мезокортикальных дофаминовых нейронов, проецирующих свои аксоны в префронтальную кору [90-92].
Разработка гипотезы гипофункции глутаматных NMDA-рецепторов позволила согласовать видимое противоречие между избыточной активностью мезолимбических дофаминовых нейронов и недостаточной активностью мезокортикальных дофаминовых нейронов [82].
У пациентов с шизофренией причина сбоя глутаматергической нейротрансмиссии может крыться в нарушениях генов [80,93,94], в аутоиммунной реакции в отношении NMDA-рецептора, либо других дефектах, вызывающих потерю функции глутаматергических нейронов, проецирующих аксоны в ствол мозга, либо же в нарушении процесса передачи сигнала NMDA-рецепторами [75-77]. Перечисленные нарушения вызывают потерю стимулирующих глутаматергических сигналов, получаемых NMDA-рецепторами тормозных интернейронов ствола мозга, что приводит к растормаживанию мезолимбических дофаминовых нейронов, дофаминовой гиперстимуляции их целевых клеток, и, как следствие, к позитивным психотическим симптомам [82,95]. И напротив, потеря функции кортико-стволовых глутаматергических проекций и/или гипоактивность NMDA-рецепторов на поверхности мезо-кортикальных дофаминовых нейронов снижает степень тонического возбуждения данных мезокортикальных дофаминовых нейронов и их клеток-мишеней в префронтальной коре. Считается, что гипоактивность данных мезокортикальных путей связана с негативными симптомами и нарушениями когнитивных и аффективных функций [80,82-84,93,96,97].
Отмеченная нами флуктуация уровней аутоантител к FRa в сыворотке крови у пациентов с резистентной шизофренией хорошо согласуется с гипотезой гипофункции NMDA-рецептора. Связь шизофрении с нарушениями метаболизма фолата и одноуглеродного метаболизма подтверждается также сообщениями о случаях врожденной недостаточности фермента MTHFR, нарушений переноса одноуглеродных фрагментов, а также об улучшении состояния пациентов после назначения фолата либо (DL)- 5-метилтетрагидрофолата [98-101].
Во время фазы, при которой титры аутоантител в сыворотке повышены, у пациента снижено поступление фолата в ЦНС – это приводит к внезапному изъятию фолата из активных реакцией промежуточного метаболизма и метаболических цепочек, зависимых от наличия фолата. Можно объяснить отмечаемые при шизофрении когнитивные нарушения и потерю серого вещества накоплением гомоцистеина, который действует как стимулирующий агонист NMDA-рецепторов, с последующей гиперстимуляцией, эксайтотоксичностью и апоптозом нейронов, содержащих NMDA-рецепторы [27, 47, 102-105]. Параллельно с этим истощение запасов фолата в мозге снижает выработку и доступность тетрагидробиоптерина (BH4), являющегося кофактором тирозин- и триптофангидроксилазы, а также нейрональной NO-синтазы. При низких уровнях внутриклеточного BH4 производство дофамина и серотонина снижается, а фермент NO-синтаза катализирует выработку не NO, а свободного радикала пероксинитрита [55-57]. Недостаточность фолата в мозге приводит к накоплению гомоцистеина и к нитрозативному стрессу в NMDAr-экспрессирующих нейронах. Ввиду этих негативных метаболических изменений данная группа нейронов становится особенно уязвимой к перепроизводству NO, за которым следует нарушение функции клетки и запускается апоптоз [106, 107]. Прогрессирующая потеря NMDAr-экспрессирующих нейронов может послужить объяснением отмечаемой при сканировании мозга медленной потере серого вещества, так же как и потере ГАМКергических интернейронов, связанных с мезолимбическими дофаминовыми нейронами ствола мозга, которая должна приводить к расторможению дофаминергической стимуляции стриато-лимбических мишеней и вызывать позитивные симптомы. Потеря кортико-стволовых глутаматергических проекций, ведущих к мезокортикальным дофаминергическим нейронам, либо апоптоз данных дофаминовых нейронов из-за токсического воздействия гомоцистеина либо нитрозативного стресса предрасполагает данные мезокортикальные пути к сниженной активности – этим можно объяснить возникновение негативных симптомов и снижение когнитивных способностей.
При снижении либо исчезновении аутоантител к FRa приток фолата к нейронам восстанавливается, вновь запуская фолат-зависимые нейрометаболические процессы. Следующий за этим всплеск уровней дофамина и серотонина может привести к относительной гиперстимуляции соответствующих рецепторов, экспрессированных на низком уровне, и способствовать тем самым развитию позитивных симптомов.
6. Фолат и депрессия
16 взрослых пациентов (средний возраст±СКО: 48.3±19.5 лет; диапазон: 17-85 лет) с большим депрессивным расстройством, резистентным к терапии, были проверены на наличие блокирующих аутоантител к FRa. У семерых пациентов из шестнадцати аутоантитела к FRa в сыворотке отсутствовали, у 9 аутоантитела были обнаружены, причем у большинства из них титр был довольно низок (среднее±СКО: 0.28±0.52 пмоль блокированных FRa/мл сыворотки; диапазон: 0-1.95), за исключением двух пациентов, у которых были отмечены высокие титры антител. У одной 85-летней женщины, более 9 лет страдавшей резистентной большой депрессией, был отмечен очень высокий уровень аутоантител – выше 1.95 пмоль блокированного FRa/мл сыворотки. У второй пациентки, 29-летней женщины, страдавшей аутоиммунным тиреоидитом и послеродовой депрессией, изначально наблюдался высокий уровень аутоантител к FRa, но с исчезновением аутоантител её депрессия спонтанно прекратилась. У её сына на фоне нормального речевого и умственного развития развился синдром спастической атаксии из-за ЦФН, ассоциированной с аутоантителами к FRa. В большом количестве публикаций сообщается, что у некоторых пациентов с большим депрессивным расстройством назначение фолата в качестве дополнительной терапии даёт положительные результаты [108-114].
7. Терапевтические стратегии
Выбор терапии при каждой из разновидностей синдрома ЦФН основывается на клиническом диагнозе и биохимических исследованиях: анализе спинномозговой жидкости, поиске аутоантител к FRa в сыворотке крови, диагностике возможных митохондриальных заболеваний и дефектов гена FRa.
У пациентов раннего детского возраста терапия младенческой формы ЦФН, аутизма с неврологическими отклонениями и синдрома спастической атаксии с ЦФН, вызванных аутоантителами к FRa, проводится назначением фолиниевой кислоты в большой дозировке (0.5-1 мг/кг/сутки). Такие дозы фолиниевой кислоты достаточны, чтобы повысить уровни фолата в сыворотке. Фолиниевая кислота способна преодолевать гематоэнцефалический барьер с помощью низкоаффинного переносчика восстановленных фолатов-1 (RFC1), открывающего альтернативный путь доступа в спинномозговую жидкость. Если назначение фолиниевой кислоты в дозировке 0.5-1 мг/кг не приводит к клиническому улучшению, возможно постепенное увеличение дозировки. При дефектах гена FRa обычно требуется более высокая дневная доза фолиниевой кислоты – от 2 до 5 мг/кг.
Большинство препаратов фолиниевой кислоты, доступных на рынке, представляют собой DL-рацемат 5-формилтетрагидрофолата, но существует и препарат, в составе которого только природный L-стереоизомер кислоты. Учитывая то, что DL-фолиниевая кислота назначается в высоких дозах, а неестественный изомер долго выводится из организма, было сделано предположение, что он способен напрямую ингибировать фолат-зависимые ферменты, такие как тимидилатсинтаза [115]. Следует избегать назначения больших доз фолиевой кислоты, поскольку при использовании больших доз этот полностью окисленный фолат способен проникать сквозь гематоэнцефалический барьер и замещать метаболически активные восстановленные формы фолата. Считается, что после захвата фолиевой кислоты нейроном она не может быть восстановлена до тетрагидрофолата внутри клетки, потому что нейрональная дигидрофолатредуктаза человека, согласно исследованиям, крайне малоактивна, однако в недавнем исследовании была продемонстрирована нормальная экспрессия дигидрофолатредуктазы в тканях мозга эмбрионов и взрослых людей [116, 117]. Доступные в продаже препараты L-метилтетрагидрофолата пока что не были исследованы при синдромах ЦФН у детей, но L-метилтетрагидрофолат был успешно применен в исследованиях взрослых пациентов с большим депрессивным расстройством [108].
Молочные продукты животного происхождения содержат растворимый FR, почти идентичный по структуре человеческому антигену FRa [72]. У лиц с генетической предрасположенностью растворимый FR провоцирует генерацию FRa-специфичных аутоантител, атакующих FRa на клетках сосудистого сплетения. Было показано, что полный отказ от употребления молока животных приводит к значительному снижению титров аутоантител к FRa, что согласуется с результатами исследований детей с аутизмом, в которых было показано, что у некоторых из них переход на безглютеновую и безмолочную диету приводит к улучшению состояния [118,119]. Более того, коррекцией режима питания можно снизить либо предотвратить колебания титров аутоантител к FRa при аутизме, шизофрении и шизоаффективных расстройствах [72]. Есть опыт использования кортикостероидов, иммуноглобулинов и иммунодепрессантов при синдромах ЦФН, вызванных аутоиммунной реакцией на FRa, однако эти препараты не должны рассматриваться в качестве терапии первой линии, поскольку их применение связано с риском развития серьезных нежелательных явлений.
Было отмечено, что терапия фолиниевой кислотой дает великолепные результаты в случаях, когда диагноз ставится быстро и препарат назначается сразу после появления первых признаков и симптомов заболевания у пациентов с младенческой формой ЦФН либо с аутизмом с неврологическими отклонениями и наличием аутоантител к FRa. Прогноз ухудшается при увеличении временного промежутка между появлением симптомов и постановкой диагноза/началом лечения [36]. В связи с этим следует рекомендовать скрининг на аутоантитела к FRa у пациентов с дебютом симптомов либо с подозрением на синдром ЦФН.
8. Подход к диагностике
В научной литературе описана картина младенческого синдрома ЦФН – орфанного заболевания, симптомы которого становятся заметны в возрасте 4-6 месяцев. При подозрении на младенческий синдром ЦФН спинномозговая жидкость ребенка должна быть проанализирована на возможное снижение уровней MTHF. После подтверждения сниженного уровня MTHF в ликворе следует проанализировать сыворотку на наличие аутоантител к FRa, осуществить секвенирование гена FRa, провести диагностику митохондриальных заболеваний. Чем раньше будет назначена терапия большими дозами фолиниевой кислоты, тем благоприятней будет прогноз. В научной литературе приведена схема диагностического обследования, включающая дифференциальную диагностику и руководство по терапии [16]. Факт большой распространенности (55-59%) блокирующих аутоантител к FRa среди пациентов с низкофункциональным аутизмом говорит о необходимости проведения анализа на наличие данных аутоантител, поскольку назначение фолиниевой кислоты и безмолочной диеты, возможно, способно улучшить состояние большинства пациентов.
Диагностическое и статистическое руководство по психическим расстройствам (DSM-5), разработанное с учетом мнений специалистов всего мира, может использоваться для первоначальной диагностики: этот документ содержит критерии диагностики отдельных заболеваний, таких как шизофрения и большое депрессивное расстройство. При диагностике шизофрении проводится сбор подробного анамнеза пациента и членов его семьи – при этом должны быть учтены вредные привычки пациента (употребление каннабиса, кокаина, опиоидов, психостимуляторов, галлюциногенов) и осуществлено психометрическое тестирование для оценки IQ и когнитивных способностей. Принимается во внимание наличие сопутствующих заболеваний и характер терапии (препаратов), назначенной по их поводу. Шизофрению следует дифференцировать от схожих психиатрических состояний, таких как биполярно-аффективное либо шизоаффективное расстройство. Существуют специальные диагностические шкалы для оценки симптомов шизофрении, например, Шкала оценки позитивных и негативных синдромов (PANSS). При первом подозрении на шизофрению, сделанном на основании клинических критериев, следует провести диагностическое исследование, включающее сканирование мозга, анализ электроэнцефалограммы и дифференциальную лабораторную диагностику. В ходе диагностического исследования у пациента исключаются состояния, мимикрирующие под шизофрению. Список диагнозов, учитываемых при дифференциальной диагностике, включает анатомические повреждения (например, опухоли мозга), редкие врождённые нарушения метаболизма и болезни накопления метаболитов (порфирия, GM2-ганглиозидоз, метахроматическая лейкодистрофия, адренолейкодистрофия), эндокринные заболевания (тяжелый гипо- и гипертиреоз, гипо- и гиперпаратиреоз), отравление тяжелыми металлами (свинцом, марганцем), инфекционные и пара-инфекционные заболевания (нейросифилис, нейроборрелиоз), эпилепсию височных долей, деменцию, аутоиммунные состояния (сифилис, рассеянный склероз, либмический энцефалит с анти-NMDAr-антителами), и недостаток некоторых витаминов (B12, тиамин, рибофлавин). После того, как завершился процесс постановки диагноза шизофрении с исключением схожих состояний, начинается долгий процесс психотерапевтической и психосоциальной реабилитации пациента, обычно в сочетании с нейролептиком, призванным заглушить позитивные симптомы.
Терапия нейролептиками в сочетании с психотерапией дает эффект примерно у 80% пациентов с шизофренией, а у примерно 20% пациентов даже при адекватном терапевтическом вмешательстве на протяжении 1 года не удается добиться улучшений либо эти улучшения недостаточны. Среди таких пациентов, страдающих резистентной шизофренией, распространенность аутоиммунных реакций в отношении к FRa может достигать 85%. Поскольку титры антител к FRa в сыворотке крови могут претерпевать значительные колебания, мы рекомендуем в целях исключения анти-FRa аутоиммунного расстройства анализировать как минимум три образца сыворотки, собранных с недельными интервалами. Что касается пациентов с большим депрессивным расстройством, то полученные нами предварительные результаты говорят о целесообразности отбора для анализа на наличие аутоантител к FRa пациентов с резистентностью к терапии, у которых, согласно оценкам, антитела обнаруживаются в 56% случаев. Однако следует отметить, что полученные нами данные требуют подтверждения в последующих исследованиях, целью которых должно быть определение исходной распространенности антител к FRa в разных группах пациентов, страдающих шизофренией либо большим депрессивным расстройством.
Список литературы
- Reynolds, Vitamin B12, folic acid, and the nervous system, Lancet Neurol. 5 (2006) 949-60.
- D. Shorvon, M.W.P. Carney, I. Chanarin, E.H. Reynolds, The neuropsychiatry of megaloblastic anaemia, BMJ 281 (1980) 1036–38.
- G. Lever, R.D. Elwes, A. Williams, E.H. Reynolds, Subacute combined degeneration of the spinal cord due to folate deficiency: response to methyl folate treatment, J.Neurol.Neurosurg Psychiatry 49(10) (1986) 1203-7.
- H. Reynolds, The neurology of folic acid deficiency, Handb. Clin. Neurol. 120 (2014) 927-43.
- D. Shorvon, E.H. Reynolds, Folate deficiency and peripheral neuropathy, in: M.I. Botez, E.H. Reynolds (Eds), Folic acid in neurology, psychiatry and internal medicine, Raven Press, New York, 1979, pp. 413–21.
- J. Lewis,, D.A. Lawlor, G. Davey Smith, et al, The thermolabile variant of MTHFR is associated with depression in the British Women’s Heart and Health Study and a meta-analysis, Mol Psychiatry 11 (2006) 352–60.
- I. Ramos, L.H. Allen, D.M. Mungas, et al, Low folate status is associated with impaired cognitive function and dementia in the Sacramento area Latino study on aging, Am. J. Clin. Nutr. 82 (2005) 1346–52.
- Sneath, I. Chanarin, H.M. Hodkinson, C.K. McPhearson, E.H. Reynolds, Folate status in a geriatric population and its relation to dementia, Age Ageing 2 (1973) 177–82.
- H. Reynolds. Folic acid, ageing, depression, and dementia, BMJ 324 (2002) 1512–15.
- S. Goodwin, J.M. Goodwin, P.J. Garry, Association between nutritional status and cognitive functioning in a healthy elderly population, JAMA 249 (1983) 2917–21.
- I. Botez, F. Fontaine, T. Botez, J. Bachevalier, Folate-responsive neurological and mental disorders: report of 16 cases, Eur. Neurol. 16 (1977) 230–46.
- Corbeel, G. Van den Berghe,, J. Jaeken, J. Van Tornout, R. Eeckels, Congenital folate malabsorption, Eur. J. Pediatr. 143 (1985) 284 – 90.
- Qiu, M. Jansen, A. Sakaris, S.H. Min, S. Chattopadhyay, E. Tsai, et al, Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption, Cell 127 (2006) 917 – 28.
- T. Ramaekers, M. Häusler, T. Opladen, G. Heimann, N. Blau, Psychomotor retardation, spastic paraplegia, cerebellar ataxia and dyskinesia associated with low 5-methyltetrahydrofolate in cerebrospinal fluid: a novel neurometabolic condition responding to folinic acid substitution, Neuropediatrics 33 (2002) 301 – 8.
- T. Ramaekers, N. Blau, Cerebral folate deficiency, Dev. Med. Child Neurol. 46 (2004) 843 – 51.
- T. Ramaekers, J.M. Sequeira, E.V. Quadros, Clinical recognition and aspects of cerebral folate deficiencies, Clinical Chemistry and Laboratory Medicine 51 (2013) 545-554.
- T. Ramaekers, S.P. Rothenberg, J.M. Sequeira, T. Opladen, N. Blau, E.V. Quadros, et al, Autoantibodies to folate receptors in the cerebral folate deficiency syndrome, N. Engl. J. Med. 352 (2005) 1985 – 91.
- P. Rothenberg, M.P. da Costa, J.M. Sequeira, J. Cracco, J.L. Roberts, J. Weedon, et al, Autoantibodies against folate receptors in women with a pregnancy complicated by a neural-tube defect, N. Engl. J. Med. 350 (2004)134 – 42.
- Grapp, A.Wrede, M. Schweizer, S. Hüwel, H.J. Galla, N. Snaidero, M. Simons, J. Bückers, P.S. Low, H. Urlaub, Gärtner, R. Steinfeld, Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma, Nat. Commun. 4 (2013) 2123.
- Steinfeld, M. Grapp, R. Kraetzner, S. Dreha-Kulaczewski, G. Helms, P. Dechent, R. Wevers, S. Grosso, J. Gärtner, Folate receptor alpha defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism, Am. J. Hum. Genet., 85 (3) (2009) 354-63.
- Grapp, I.A. Just, T. Linnankivi, P. Wolf, T. Lücke, M. Häusler, J. Gärtner, R. Steinfeld, Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency, Brain 135 (2012) 2022-31.
- T. Ramaekers, J. Weis, J.M. Sequeira, E.V. Quadros, N. Blau, Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency, Neuropediatrics 38 (2007) 184 – 7.
- Garcia-Cazorla, E.V. Quadros, A. Nascimento, M.T. Garcia-Silva, P. Briones, J. Montoya, et al, Mitochondrial diseases associated with cerebral folate deficiency, Neurology 70 (2008) 1360 – 2.
- Tanji, E.A. Schon, S. DiMauro, E. Bonilla, Kearns-Sayre syndrome: oncocytic transformation of choroid plexus epithelium, J. Neurol. Sci. 178 (2000) 29 – 36.
- T. Ramaekers, S.I. Hansen, J. Holm, T. Opladen, J. Senderek, M. H äusler, et al, Reduced folate transport to the CNS in female Rett patients, Neurology 61 (2003) 506 – 15.
- T. Ramaekers, J.M. Sequeira, R. Artuch, N. Blau, T. Temudo, A. Ormazabal A, et al, Folate receptor autoantibodies and spinal fluid 5-methyltetrahydrofolate deficiency in Rett syndrome, Neuropediatrics 38 (2007) 179 – 83.
- T. Ramaekers, B. Thöny, J.M. Sequeira, M. Ansseau, P. Philippe, F. Boemer, V. Bours, E.V. Quadros, Folinic acid treatment for schizophrenia associated with folate receptor autoantibodies, Mol. Genet. Metab. 113 (4) (2014) 307-14.
- E. Frye, J.M. Sequeira, E. Quadros, D. Rossignol, Folate receptor alpha autoantibodies modulate thyroid function in autism spectrum disorder, N.A.J. Med. Sci. 7(2) (2014) 53-56.
- A. Piedrahita, B. Oetama, G. Bennett, J. van Waes, B.A. Kamen, J. Richardson, et al, Mice lacking the folic acid- binding protein Folbp1 are defective in early embryonic development, Nat. Genet. 23 (1999) 228 – 32.
- Spiegelstein, L.E. Mitchell, M.Y. Merriweather, N.J. Wicker, Q. Zhang, E.J. Lammer, R.H. Finnell, Embryonic development of folate binding protein-1 (Folbp1) knockout mice: Effects of the chemical form, dose, and timing of maternal folate supplementation, Dev. Dyn. 231 (1) (2004) 221-31.
- H. Finnell, A. Gould, O. Spiegelstein, Pathobiology and genetics of neural tube defects, Epilepsia 44 (2003) Suppl 3: 14-23. Review.
- J. Schmidt, D.J. Tancredi, S. Ozonoff, R.L. Hansen, J. Hartiala, H. Allayee, et al, Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (Childhood Autism Risks from Genetics and Environment) case-control study, Am. J. Clin. Nutr. 96 (2012) 80 – 9.
- Surén, C. Roth, M. Bresnahan, M. Haugen, M. Hornig, D. Hirtz, K.K. Lie, W.I. Lipkin, P. Magnus, T. Reichborn- Kjennerud, S. Schjølberg, G. Davey Smith, A.S. Øyen, E. Susser, C. Stoltenberg. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children, JAMA 309 (6) (2013) 570-7.
- T. Ramaekers, E.V. Quadros, J.M. Sequeira, Role of folate receptor autoantibodies in infantile autism, Mol. Psychiatry 18 (3) (2013) 270-1.
- Shapira, J.M. Sequeira, E.V. Quadros, Folate receptor autoantibodies in pregnancy related complications, Birth Defects Res. A. Clin. Mol. Teratol. (2015) Sep 21. doi: 10.1002/bdra.23436. [Epub ahead of print] PubMed PMID: 26390016.
- T. Ramaekers, N. Blau, J.M. Sequeira, M.C. Nassogne, E.V. Quadros, Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits, Neuropediatrics 38 (2007) 276 – 81.
- Moretti, T. Sahoo, K. Hyland, T. Bottiglieri,S. Peters, D. del Gaudio, et al, Cerebral folate deficiency with developmental delay, autism, and response to folinic acid, Neurology 64 (2005) 1088 – 90.
- E. Frye, J.M. Sequeira, E.V. Quadros, S.J. James, D.A. Rossignol, Cerebral folate receptor autoantibodies in autism spectrum disorder, Mol. Psychiatry. 18 (3) (2013) 369-81.
- J. Hansen, N. Blau, Cerebral folate deficiency: life-changing supplementation with folinic acid, Mol. Genet. Metab. 84 (2005) 371 – 3.
- Hasselmann, N. Blau, V.T. Ramaekers, E.V. Quadros, J.M. Sequeira, M. Weissert, Cerebral folate deficiency and CNS inflammatory markers in Alpers disease, Mol. Genet. Metab. 99 (2010) 58 – 61.
- U. Steele, S.M. Cheah, A. Veerapandiyan, W. Gallentine, E.C. Smith, M.A. Mikati,.Electroencephalographic and seizure manifestations in two patients with folate receptor autoimmune antibody-mediated primary cerebral folate deficiency, Epilepsy Behav. 24(4) (2012) 507-12.
- Gordon, Cerebral folate deficiency, Dev. Med. Child Neurol. 51(3) (2009) 180-2.
- Ramaekers, E.V. Quadros, Folate receptor autoimmunity in cerebral folate deficiency, in: R.C. Dale, A. Vincent, (Eds.), Inflammatory and autoimmune disorders of the nervous system in children, Mac Keith Press, London, 2010, pp.302-15.
- Zhao, N. Diop-Bove, M. Visentin, I.D. Goldman, Mechanisms of membrane transport of folates into cells and across Epithelia, Annu. Rev. Nutr. 31 (2011) 177-201.
- J. Bagley, J. Selhub J, A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells, Proc. Natl. Acad. Sci. USA 95 (1998) 13217-13220.
- T. Clayton, I. Smith, B. Harding, K. Hyland, J.V. Leonard, R.J. Leeming, Subacute combined degeneration of the cord, dementia and Parkinsonism due to an inborn error of folate metabolism, J.N.N.P. 49 (1986) 920-927.
- Obeid, W. Herrmann, Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia, F.E.B.S. Letters 580 (2006) 2994-3005.
- Obeid, P. Kostopoulos, J.P. Knapp, M. Kasoha, G. Becker, K. Fassbender, W. Herrmann, L. Schaevitz, Biomarkers of Folate and Vitamin B12 Are Related in Blood and Cerebrospinal Fluid, Clinical Chemistry 53 (2) (2007) 326-333.
- Bottiglieri, M. Laundy, R. Crellin, B.K. Toone, M.W. Carney, E.H. Reynolds, Homocysteine, folate, methylation, and monoamine metabolism in depression, J. Neurol. Neurosurg. Psychiatry 69(2) (2000) 228-32.
- R. Schaevitz, J.E. Berger-Sweeney, Gene-environment interactions and epigenetic pathways in autism: the importance of one-carbon metabolism, I.L.A.R.J. 53 (3-4) (2012) 322-40.
- Opladen, N. Blau, V.T. Ramaekers, Effect of antiepileptic drugs and reactive oxygen species on folate receptor 1 (FOLR1)-dependent 5-methyltetrahydrofolate transport, Mol. Genet. Metab. 101 (1) (2010) 48-54.
- B. Aylett, V. Neergheen, I.P. Hargreaves, S. Eaton, J.M. Land, S. Rahman, S.J. Heales, Levels of 5-methyltetrahydrofolate and ascorbic acid in cerebrospinal fluid are correlated: implications for the accelerated degradation of folate by reactive oxygen species, Neurochem. Int. 63(8) (2013) 750-5.
- A. Lipton, W.K. Kim, Y.B. Choi, S. Kumar, D.M. D'Emilia, P.V. Rayudu, D.R. Arnelle, J.S. Stamler, Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor, Proc. Natl. Acad. Sci. USA 94 (1997) 5923–5928.
- R.Yang, K.A. Svensson, Allosteric modulation of NMDA receptor via elevation of brain glycine and D-serine: the therapeutic potentials for schizophrenia, Pharmacology & therapeutics 120 (2008) 317–332.
- Delgado-Esteban, A. Almeida, J.M. Medina, Tetrahydrobiopterin deficiency increases neuronal vulnerability to hypoxia, J. Neurochem. 82(5) (2002) 1148-59.
- L.Kim, Y.S. Park, Maintenance of cellular tetrahydrobiopterin homeostasis, B.M.B. Rep. 43(9) (2010) 584-92.
- R. Werner, N. Blau, B.Thöny, Tetrahydrobiopterin: biochemistry and pathophysiology, Biochem. J. 438 (3) (2011) 397-414.
- Blanchard-Fillion, J.M. Souza, T.Friel, G.C. Jiang, K.Vrana, V. Sharov, C. Barrón L,Schöneich, C. Quijano,
- Alvarez, R. Radi, S. Przedborski, G.S. Fernando, J. Horwitz, H. Ischiropoulos, Nitration and inactivation of tyrosine hydroxylase by peroxynitrite, J. Biol. Chem. 276 (49) (2001) 46017-23.
- M. Kuhn, T.J. Geddes, Peroxynitrite inactivates tryptophan hydroxylase via sulfhydryl oxidation. Coincident nitration of enzyme tyrosyl residues has minimal impact on catalytic activity, J. Biol. Chem. 274 (42)(1999) 29726-32.
- Deth, C. Muratore, J. Benzecry, V.A. Power-Charnitsky, M. Waly, How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis, Neurotoxicology. 29 (1) (2008) 190-201.
- Lozoff, Early iron deficiency has brain and behavior effects consistent with dopaminergic dysfunction, J. Nutr. 141 (4) (2011) 740S-746S.
- E. Brunette, P.V. Tran, J.D. Wobken, E.S. Carlson, M.K. Georgieff, Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus, Dev. Neurosci. 32(3) (2010) 238-48.
- Takeda, Zinc homeostasis and functions of zinc in the brain, Biometals. 14(3-4) (2001)343-51.
- Takeda, Manganese action in brain function, Brain Res. Rev. 41(1) (2003) 79-87.
- Y. Uriu-Adams, R.E. Scherr, L. Lanoue, C.L, Keen. Influence of copper on early development: prenatal and postnatal Considerations, Biofactors 36 (2) (2010) 136-52.
- Johnson, Micronutrient accumulation and depletion in schizophrenia, epilepsy, autism and Parkinson's disease?, Med. Hypotheses. 56(5) (2001) 641-5.
- Benton, R. T. Donohoe, The effects of nutrients on mood, Public Health Nutr. 2(3A) (1999) 403-9.
- E. Frye, S.J. James, Metabolic pathology of autism in relation to redox metabolism, Biomark. Med. 8(3) (2014) 321-30.
- J. James, S. Melnyk, S. Jernigan, M.A. Cleves, C.H. Halsted, D.H. Wong, P. Cutler, K. Bock, M. Boris, J.J. Bradstreet, S.M. Baker, D.W. Gaylor, Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism, Am. J. Med. Genet. B. Neuropsychiatr. Genet. 141B (8) (2006) 947-56.
- M. Ueland, Choline and betaine in health and disease, J. Inherit. Metab. Dis.34(1) (2011) 3-15.
- Boshnjaku, K.W. Shim, T. Tsurubuchi, S. Ichi, E.V. Szany, G. Xi, B. Mania-Farnell, D.G. McLone, T. Tomita and C. Mayanil, Nuclear localization of folate receptor alpha: a new role as a transcription factor, Sci. Rep. 2 (2012) 980.
- T. Ramaekers, J.M. Sequeira, N. Blau, E.V. Quadros, A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome, Dev. Med. Child Neurol. 50 (2008) 346 – 52.
- Lambrot, C. Xu, S. Saint-Phar, G. Chountalos, T. Cohen, M. Paquet, M. Suderman, M.Hallett, S. Kimmins S, Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes, Nat. Commun., 4 (2013) 2889.
- G. Swayne, A. Kawata, N.A. Behan, A. Williams, M.G. Wade, A.J. Macfarlane, C.L. Yauk, Investigating the effects of dietary folic acid on sperm count, DNA damage and mutation in Balb/c mice, Mutat. Res. 737 (1-2) (2012) 1-7.
- S. Zandi, S.R. Irani, B. Lang, P. Waters, P.B. Jones, P. McKenna, A.J. Coles, A. Vincent, B.R. Lennox, Disease- relevant autoantibodies in first episode schizophrenia, J..Neurol. 258 (4) (2011) 686–688.
- Iizuka, F. Sakai, T. Ide, T. Monzen, S. Yoshii, M. Ligaya, K. Suzuki, D.R. Lynch, N. Suzuki, T. Hata, J. Dalmau, Anti-NMDA receptor encephalitis in Japan: long-term outcome with tumor removal, Neurology 70 (7) (2008) 504–511.
- G. Bien, A. Vincent, M.H. Barnett, A.J. Becker, I. Bluemcke, F. Graus, K.A. Jellinger, D.E. Reuss, T. Ribalta, Schlegel, I. Sutton, H. Lassmann, J. Bauer, Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis, Brain 135 (2012)1622–1638.
- H. Schultz, S.W. North, C.G. Shields, Schizophrenia: a review, Am. Fam. Physician 75 (2007) 1821–1829.
- M. Picchioni, R.M. Murray, Schizophrenia, B.M.J. 335 (2007) 91–95.
- L. Roth, Contributions of molecular biology to antipsychotic drug discovery: promises fulfilled or unfulfilled?, Dialogues Clin. Neurosci. 8 (3) (2006) 303–309.
- J. Owen, N.M. Williams, M.C. O'Donovan, The molecular genetics of schizophrenia: new findings promise new insights, Mol. Psychiatry 9 (2004) 14–27.
- M. Stahl, Beyond the dopamine hypothesis to the NMDA glutamate receptor hypofunction hypothesis of schizophrenia, C.N.S. Spectr. 4 (2007) 265–268.
- T. Coyle, NMDA receptor and schizophrenia: a brief history, Schizophr. Bull. 38 (5) (2012) 920–926.
- R. Mohn, R.R. Gainetdinov, M.G. Caron, B.H. Koller, Mice with reduced NMDA receptor expression display behaviors related to schizophrenia, Cell 98 (1999) 427–436.
- M. Thompson, C. Vidal, J.N. Giedd, P. Gochman, J. Blumenthal, R.Nicolson, A.W. Toga, J.L.Rapoport, Mapping adolescent brain change reveals dynamic wave of accelerating grey matter loss in very early-onset schizophrenia, P.N.A.S. 98 (2001) 11650–11655.
- I. Lidsky, Reevaluation of the mesolimbic hypothesis of antipsychotic drug action, Schizophr. Bull. 21(1) (1995) 67-74. Review.
- Lamelle, A. Abi-Dargham, Dopamine in the history of the schizophrenic brain: recent contributions of brain-imaging Studies, Dialogues Clin. Neurosci. 2(4) (2000) 359-72.
- Ginovart, S. Kapur, Role of dopamine D(2) receptors for antipsychotic activity, Handb. Exp. Pharmacol.. 212 (2012) 27-52.
- T. Coyle, D. Balu, M. Benneyworth, A. Basu, A. Roseman, Beyond the dopamine receptor: novel therapeutic targets for treating schizophrenia, Dialogues Clin. Neurosci. 12(3) (2010) 359-82.
- K. Seamans, C.R. Yang, The principal features and mechanisms of dopamine modulation in the prefrontal cortex, Prog. Neurobiol. 74(1) (2004) 1-58.
- D. Selemon, N. Zecevic, Schizophrenia: a tale of two critical periods for prefrontal cortical development, Transl. Psychiatry. 5 (2015) e623.
- B. Knable, D.R. Weinberger, Dopamine, the prefrontal cortex and schizophrenia, J. Psychopharmacol.11(2)(1997)123- 31.
- E. Timms, M.O. Dorschne, J. Wechsler, K.Y. Choi, R. Kirkwood, S. Girirajan, C. Baker, E.E. Eichler, O. Korvatska, K.W. Roche, M.S. Horwitz, D.W. Tsuang, Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families, J.A.M.A. Psychiatry 70(6) (2013) 582-90.
- L. Schwartz, S. Sachdeva, S.M. Stahl, Genetic data supporting the NMDA glutamate receptor hypothesis for Schizophrenia, Curr. Pharm. Des. 18(12)(2012) 1580-92.
- Kehrer, N. Maziashvili, T. Dugladze, T. Gloveli, Altered Excitatory-Inhibitory Balance in the NMDA-Hypofunction Model of Schizophrenia, Front. Mol. Neurosci. 1 (2008) 6.
- González-Maeso, S.C. Sealfon, Psychedelics and schizophrenia, Trends Neurosci. 32 (4) (2009) 225–232.
- J. Gandal, R.J. Anderson, E.N. Billingslea, G.C.Carlson, T.P. Roberts, S.J. Siegel, Mice with reduced NMDA receptor expression:more consistent with autismthan schizophrenia?, Genes Brain Behav. 11 (6) (2012) 740–750.
- R. Hutto, Folate and cobalamin in psychiatric illness, Compr. Psychiatry 38 (6) (1997) 305–314.
- Regland, Schizophrenia and single-carbon metabolism, Prog. Neuropsychopharmacol. Biol. Psychiatry 29 (7) (2005) 1124–1132.
- S. Godfrey, B.K. Toone, M.W. Carney, T.G. Flynn, T.Bottiglieri, M. Laundy, I. Chanarin, E.H. Reynolds, Enhancement of recovery from psychiatric illness by methylfolate, Lancet 336 (1990) 392–395.
- M. Freeman, J.D. Finkelstein, S.H. Mudd, Folate-responsive homocystinuria and “schizophrenia”. A defect in methylation due to deficient 5,10-methylenetetrahydrofolate reductase activity, N. Engl. J. Med. 292 (10) (1975) 491– 496.
- I. Ho, D. Ortiz, E. Rogers, T.B. Shea, Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage, J. Neurosci. Res. 70(5) (2002) 694-702.
- S. McCully, Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and Inflammation, Ann. Clin. Lab. Sci. 39(3) (2009) 219-32.
- Bonfoco, D. Krainc, M. Ankarcrona, P. Nicotera, S.A. Lipton SA, Apoptosis and necrosis : two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures, Proc. Natl. Acad. Sci. USA 91 (1995) 7162-7166.
- Bonfoco, M. Leist, B. Zhivotovsky, et al, Cytoskeletal breakdown and apoptosis elicited by NO-donors in cerebellar granule cells require NMDA-receptor activation, J. Neurochem. 67 (1996) 2484-2493.
- K. Yao, S. Leonard, R.D. Reddy, Increased nitric oxide radicals in postmortem brain from patients with schizophrenia, Schizophr. Bull. 30(4) (2004) 923-34.
- F. Nasyrova, D.V. Ivashchenko, M.V. Ivanov, N.G. Neznanov, Role of nitric oxide and related molecules in schizophrenia pathogenesis: biochemical, genetic and clinical aspects, Front. Physiol. 6 (2015) 139.
- I. Papakostas, R.C. Shelton, J.M. Zajecka, T. Bottiglieri, J. Roffman, C. Cassiello, S.M. Stahl, M. Fava, Effect of adjunctive L-methylfolate 15 mg among inadequate responders to SSRIs in depressed patients who were stratified by biomarker levels and genotype: results from a randomized clinical trial, J. Clin. Psychiatry 75(8)(2014) 855-63.
- Fava, D. Mischoulon, Folate in depression: efficacy, safety, differences in formulations, and clinical issues, J. Clin. Psychiatry 70 Suppl. 5 (2009) 12-7.
- N. Young, A.M. Ghadirian, Folic acid and psychopathology, Prog. Neuropsychopharmacol. Biol. Psychiatry 13(6) (1989) 841-63.
- I. Botez, S.N. Young, J. Bachevalier, S. Gauthier, Effect of folic acid and vitamin B12 deficiencies on 5-hydroxyindoleacetic acid in human cerebrospinal fluid, Ann. Neurol. 12(5) (1982) 479-84.
- W. Roman, F.H. Bembry, L-methylfolate (Deplin®): a new medical food therapy as adjunctive treatment for Depression, Issues Ment. Health Nurs. 32(2) (2011) 142-3.
- M. Stahl, L-methylfolate: a vitamin for your monoamines, J. Clin. Psychiatry 69(9) (2008) 1352-3.
- J. Mathew, Treatment-resistant depression: recent developments and future directions, Depress. Anxiety. 25(12) (2008) 989-92.
- P. Lee, R.L. Schilsky, Inhibition of thymidylate synthase by the diastereoisomers of leucovorin, Cancer Chemother. Pharmacol. 26(4) (1990) 273-7.
- 116 D.R. Makulu, E.F. Smith, J.R. Betino, Lack of dihydrofolate reductase activity in brain tissue of mammalian species: possible implications, J. Neurochem. 21(1) (1973) 241-5.
- Banka, H.J. Blom, J. Walter, M. Aziz, J. Urquhart, C.M.Clouthier, G.I. Rice, A.P. de Brouwer, E. Hilton, G. Vassallo, Will, D.E. Smith, Y.M. Smulders, R.A. Wevers, R. Steinfeld, S. Heales, Y.J. Crow, J.N. Pelletier, S. Jones, W.G. Newman, Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency, Am. J. Hum. Genet. 88(2) (2011) 216-25.
- M. Knivsberg, K.L. Reichelt, T. Høien, M. Nødland, A randomised, controlled study of dietary intervention in autistic Syndromes, Nutr. Neurosci. 5(4) (2002) 251-61.
- Whiteley, P. Shattock, A.M. Knivsberg, A. Seim, K.L. Reichelt, L. Todd, K. Carr, M. Hooper M, Gluten- and casein- free dietary intervention for autism spectrum conditions, Front. Hum. Neurosci. 6 (2013) 344.
Благодарности
Исследования, о которых рассказывается в настоящей статье, были выполнены при поддержке Fonds National de la Recherche Scientifique Belgium (FNRS; No. 3.4.540.09.F), оказанной автору VR, и с помощью гранта Autism Speaks #8202, выданного автору EVQ. Проведение исследований было одобрено Комитетом по этике больницы города Льеж и Внутренним наблюдательным советом при медицинском центре Downstate Medical Center университета штата Нью-Йорк, США.
Пояснения к рисункам
Рисунок 1. График, демонстрирующий проявления аутоиммунной реакции в отношении FRa в зависимости от возраста, в котором начинается выработка антител.
Выработка аутоантител к FRa в организме матери во время беременности повышает риск развития дефектов нервной трубки и аутизма, а появление аутоантител к FRa в раннем послеродовом периоде предрасполагает ребенка к развитию младенческой формы синдрома ЦФН, аутизма с неврологическими отклонениями либо раннего детского аутизма. В возрасте от 1 до 2 лет развивается редкий синдром спастической атаксии, в подростковом возрасте развивается резистентная шизофрения, а во взрослом возрасте наличие аутоантител к FRa может способствовать развитию депрессии и когнитивных нарушений.
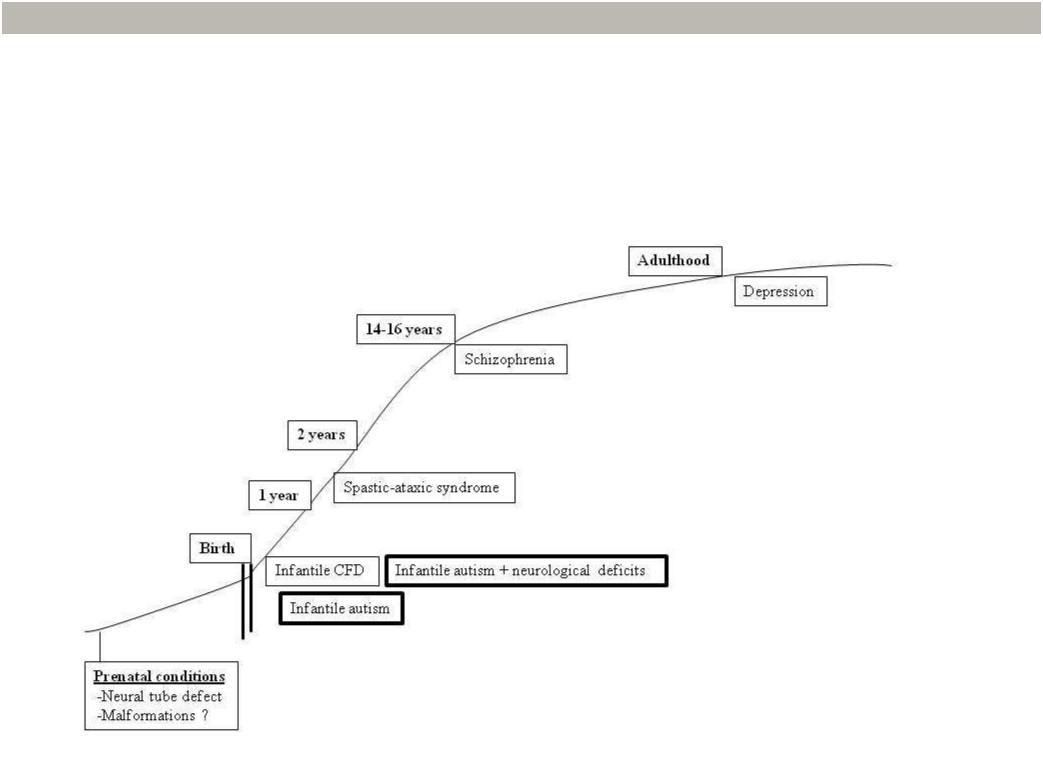
Рисунок 2. Перенос фолата через сосудистое сплетение
Перенос метилфолата через сосудистое сплетение в спинномозговую жидкость осуществляется с помощью эндоцитоза с участием FRa, расположенных на базально-латеральной поверхности эпителиальных клеток сосудистого сплетения. Перенос метилфолата из подкисленных везикул в цитоплазму, вероятно, осуществляется с помощью протон-сопряженного транспортера фолатов (PCFT). Метилфолат покидает клетку сосудистого сплетения с апикальной стороны с помощью переносчика восстановленных фолатов (RFC1). Блокирующие аутоантитела к FRa способны связываться с FRa, заякоренными на мембранах, и тем самым подавлять перенос метилфолата внутрь эпителиальных клеток. Активные формы кислорода (ROS) способны повреждать как мембранные липиды, так и белки переноса фолатов, встроенные в мембрану, и тем самым снижать общее поступление фолата в ЦНС. Активные формы кислорода также могут вступать во взаимодействие с метилфолатом непосредственно в плазме и катаболизировать его.
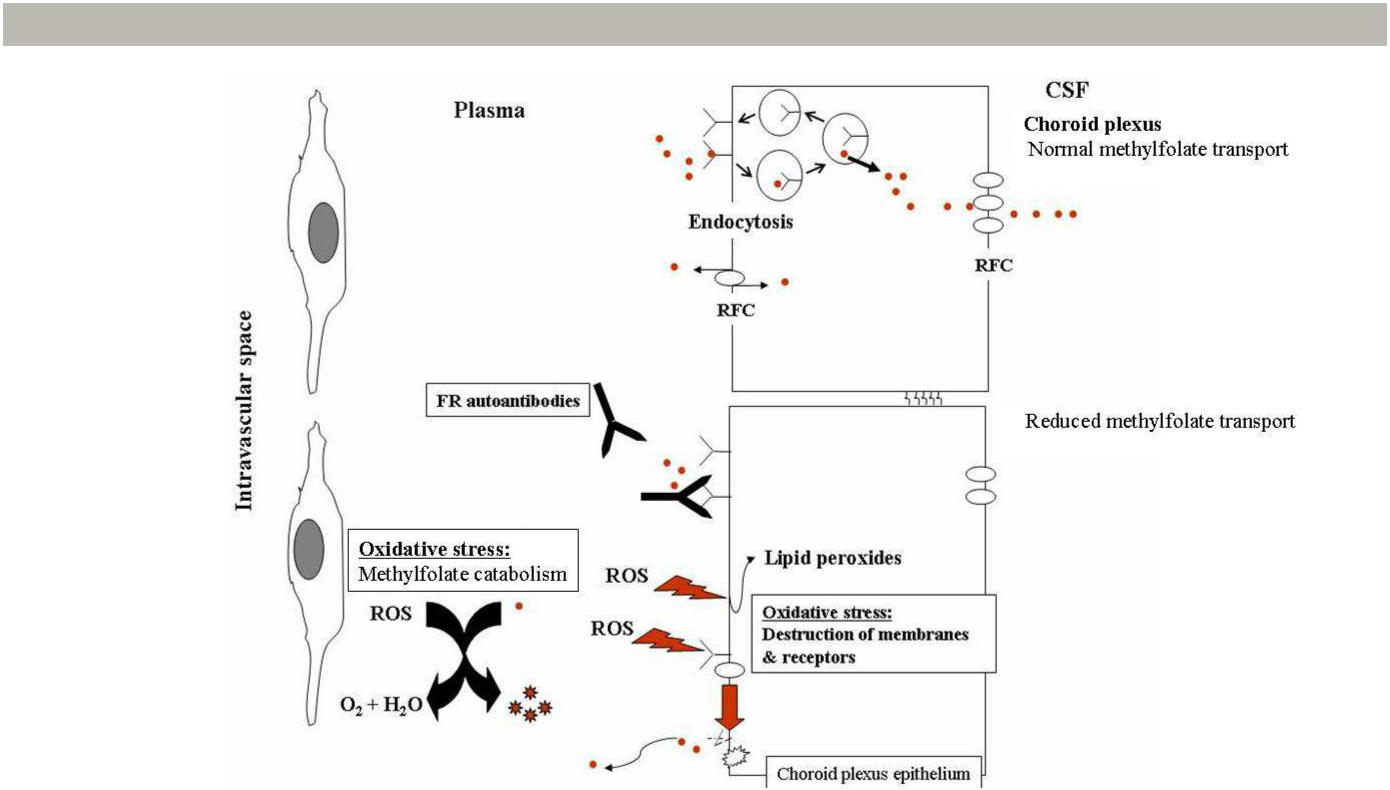
Рисунок 3. Метаболизм фолата в мозге
После FRa-опосредованного переноса N5-метилтетрагидрофолата (5-methyl THF; метилфолат) через сосудистое сплетение в ЦНС он распределяется в тканях мозга. Большая часть метилфолата, попавшая в нейроны, откладывается на хранение, и лишь небольшая доля вещества участвует в процессах метаболизма. Метилфолат отдаёт свою метильную группу B12-зависимому ферменту метионинсинтазе (MS), конвертирующему гомоцистеин в метионин. В цикле трансметилирования метионин выступает прекурсором SAM – универсального донора метильных групп, принимающего участие в более чем 100 реакциях метилирования. В цикле реметилирования тетрагидрофолат (THF) принимает моноуглеродную группу от серина и превращается в 5,10-метилентетрагидрофолат (5,10-methylene THF). Часть 5,10-метилентетрагидрофолата восстанавливается ферментом метилентетрагидрофолатредуктазой (MTHFR) до 5-метилтетрагидрофолата (5-methyl THF), а другая часть используется для синтеза тимидина либо конвертируется в 10-формилтетрагидрофолат, необходимый для синтеза пурина. В левой части рисунка: пуриновый метаболит ГТФ (GTP) выступает субстратом для фермента ГТФ-циклогидролазы I (GTPCH), синтезирующего тетрагидробиоптерин (BH4), который является кофактором ферментов, производящих дофамин, серотонин и NO. В верхней правой части рисунка изображена цепочка реакций транссульфирования. Посредством этих реакций накопленный гомоцистеин конвертируется в антиоксидант глутатион при окислительном стрессе. Сокращения: AADC: Декарбоксилаза ароматических аминокислот; Arg: аргинин; CNS: центральная нервная система; MS: метионин-синтаза; MTHFR: 5,10-метилентетрагидрофолатредуктаза; NO: оксид азота (II); NOS1: нейрональная NO-синтаза; SAM: S-аденозилметионин; SAH: S-аденозилгомоцистеин; TH: тирозингидроксилаза; TPH2: нейрональная триптофангидроксилаза; Trp: триптофан; Tyr: тирозин.
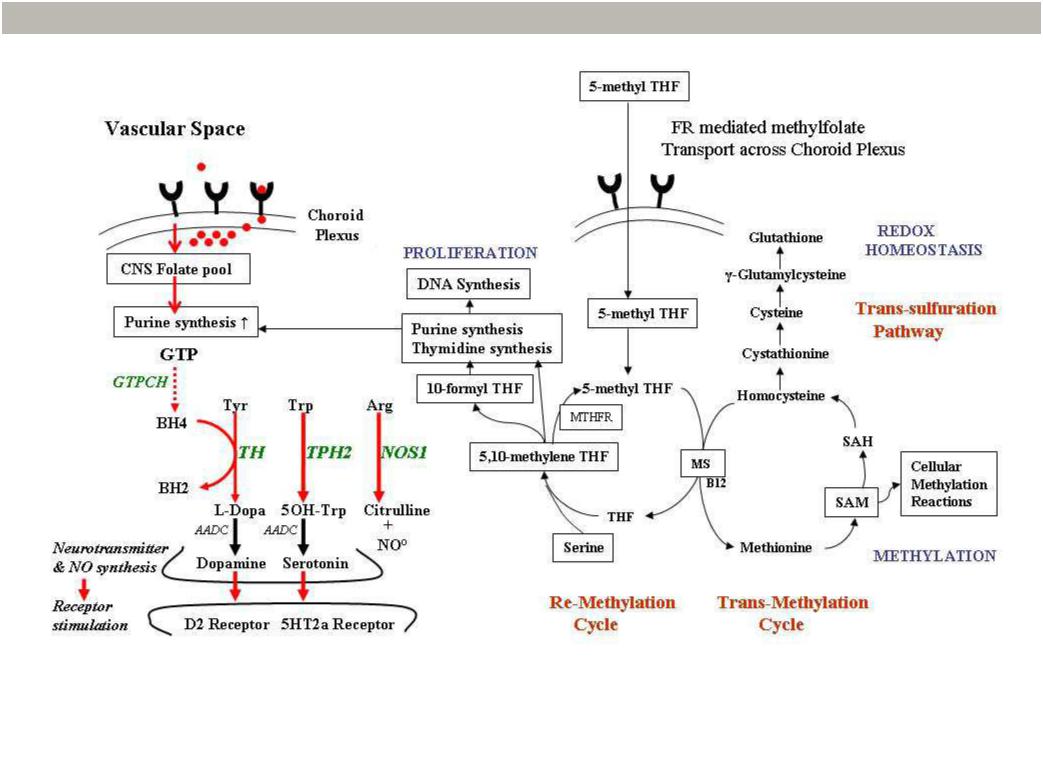
Рисунок 4. Генеалогическое древо семьи с неврологическими заболеваниями
В верхней части рисунка – нулевой пациент (женщина с шизофренией) и её младший неполнородный брат (аутизм), тети и дяди по материнской линии (LD: нарушения обучаемости, MR: умственная отсталость). В таблице в нижней части рисунка приведены уровни фолата (5MTHF) в спинномозговой жидкости, а также моноаминовых метаболитов 5-гидроксииндолуксусной кислоты (5HIAA), гомованилиновой кислоты (HVA), птериновых метаболитов неоптерина (neo) и биоптерина (bio), и сывороточных уровней аутоантител к FRa у двух пациентов до начала терапии (baseline) и после назначения фолиниевой кислоты с безмолочной диетой (folinic acid + milk free).
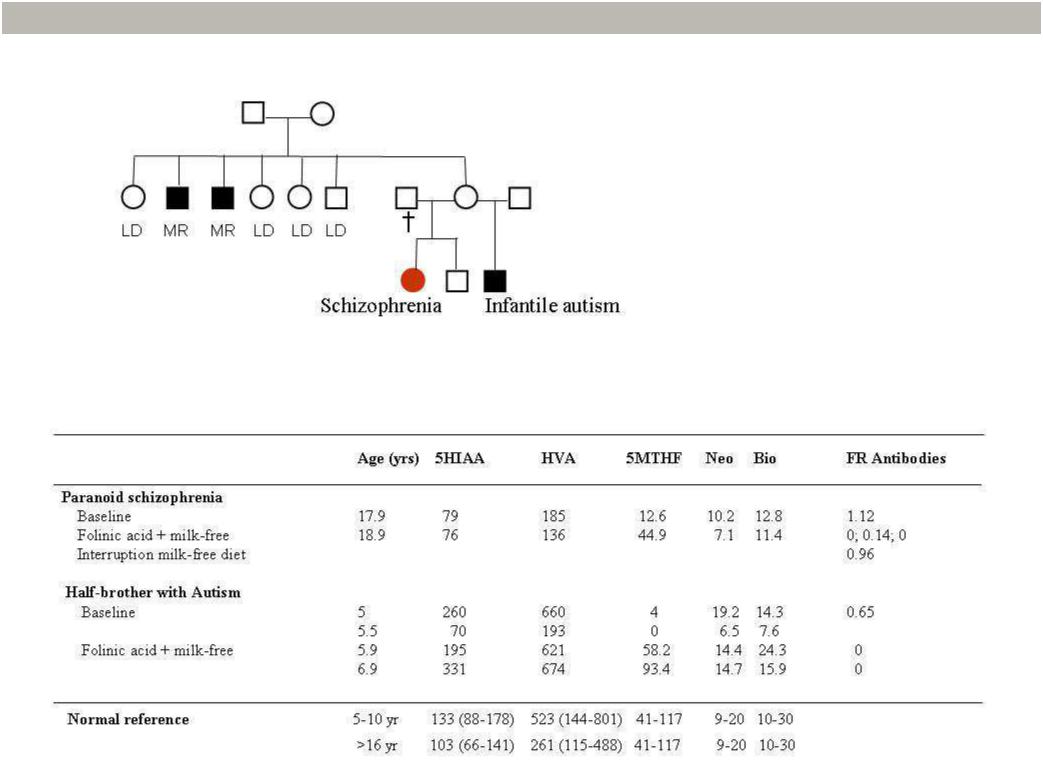
Рисунок 5. Уровни аутоантител к FRa у детей с аутизмом и их родителей
Турецкая семья с матерью-носителем блокирующих антител к FRa (вверху слева). Данная мать родила трех девочек, у каждой из которых были обнаружены антитела к FRa. У девочек был выявлен аутизм с низким IQ, а у младшей из дочерей также Ретт-подобный синдром. В другой семье (вверху посередине) антител не было обнаружено у родителей, а у их двоих сыновей был диагностирован тяжелый аутизм и выявлены аутоантитела к FRa. В тунисской семье (вверху справа) аутоантитела к FRa были обнаружены у обоих родителей, но при этом у их детей – монозиготных близнецов, страдавших тяжелой формой аутизма – анализ не выявил антител. Внизу справа - древо семьи, в которой у здорового отца с антителами к FRa родились сыновья-двойняшки с антителами, при этом у матери антитела отсутствовали.
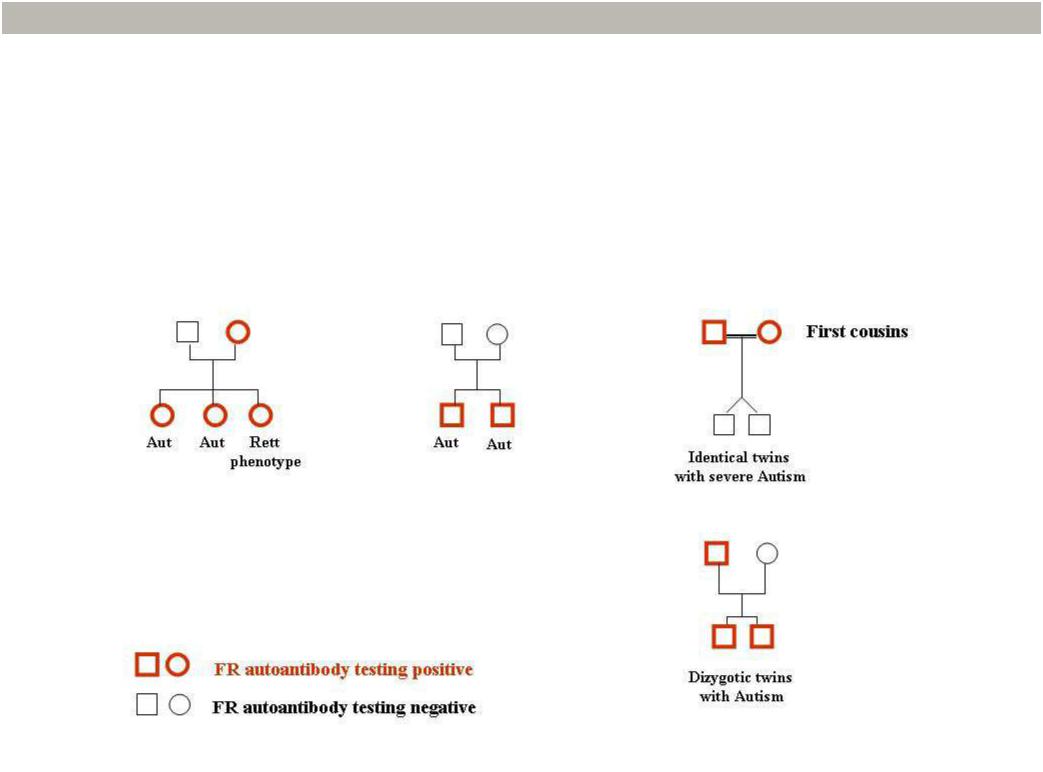
Рисунок 6. Воздействие аутоантител к FRa, блокирующих перенос фолата, на метаболизм
Аутоантитела к FRa предотвращают связывание метилфолата с FRa, присоединенными к клеточным мембранам, в результате чего снижается запас фолата в ЦНС. Снижение доступности метаболитов фолата замедляет de novo-синтез пурина, снижая производство гуанозинтрифосфата (GTP), являющегося субстратом фермента ГТФ-циклогидролазы I (GTPCH). Вследствие этого снижается скорость преобразования ГТФ в тетрагидробиоптерин (BH4), общий кофактор для тирозингидроксилазы (TH), триптофангидроксилазы (TPH2) и нейрональной NO-синтазы (NOS1) – ферментов, катализирующих синтез дофамина, серотонина и NO соответственно. Происходящее одновременно с этим накопление гомоцистеина, вызывающего гиперстимуляцию NMDA-рецепторов, а также пероксинитрита (ONOOH), формирующегося в условиях недостатка BH4, вызывает апоптоз NMDAr-экспрессирующих нейронов. Согласно гипотезе, повышение уровней антител к FR проявляется негативной симптоматикой.
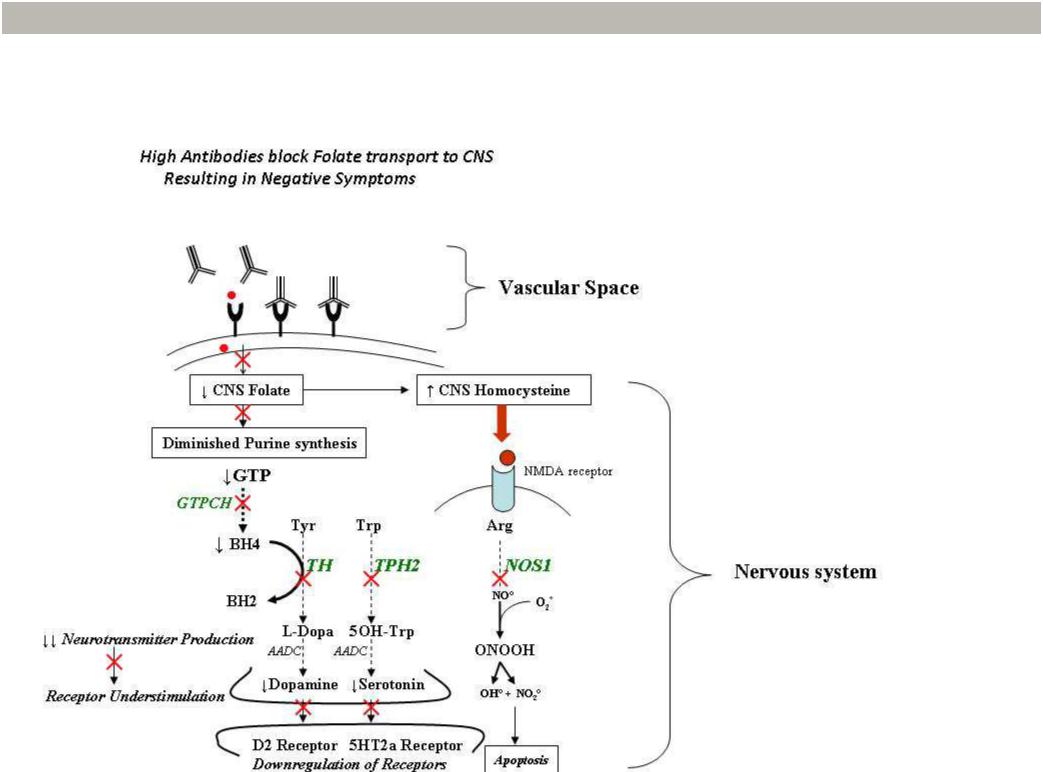
Таблица 1. Уровни фолатов и аутоантител к FRa у пациентов с нейропсихиатрическими заболеваниями
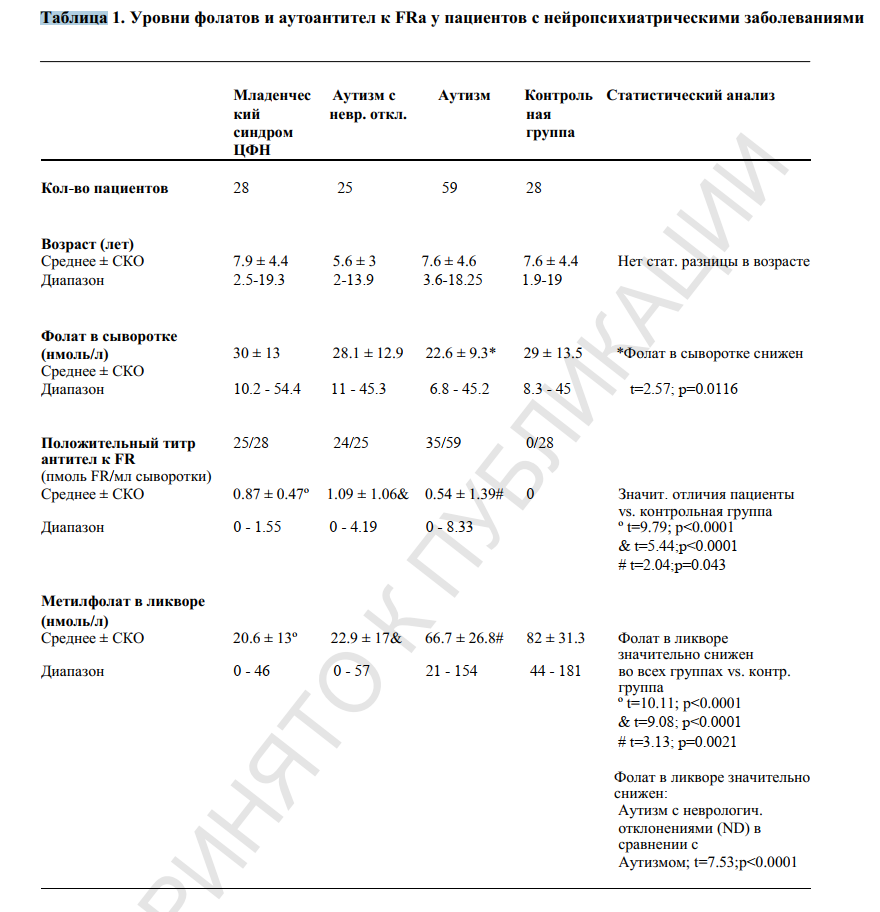
Тезисы:
Нарушения развития и нейропсихиатрические заболевания возникают под воздействием множества факторов.
У некоторых пациентов под воздействием аутоантител к фолатному рецептору альфа развивается церебральная фолатная недостаточность.
Аутоантитела к фолатному рецептору были обнаружены у большинства детей с аутизмом и шизофренией.
Была доказана полезность применения фолиниевой кислоты при данных заболеваниях.
Научная публикация: The basis for folinic acid treatment in neuro-psychiatric disorders, by V.T. Ramaekers, MD PhD, Prof Dr., J.M. Sequeira, E.V. Quadros
Немного критики, чтобы показать, что гипотеза группы Ramaekers et al пока что не является полностью доказанной. Из обзорной статьи по церебральной фолатной недостаточности, опубликованной в 2019 году в Journal of Inherited Metabolic Disease группой Pope et al.:
"Вероятно, список известных нам неврологических состояний, приводящих к ЦФН, еще неполон, поскольку у некоторых пациентов очень низкие уровни 5-MTHF в ликворе отмечаются в отсутствие какого-либо генетического диагноза. Утверждается, что аутоантитела к фолатному рецептору в ликворе пациента способны приводить к развитию ЦФН при множестве состояний – от синдрома ЦФН с дебютом в младенческом возрасте (Ramaekers et al, 2005) до шизофрении, расстройств аутистического спектра и синдрома Ретта (Ramaekers et a. 2014; Frye 2013; Moretti 2008; Ramaekers et al, 2007c and 2003). Результаты, на которых основываются эти утверждения, пока что не были реплицированы в других лабораториях. Возможно, что при некоторых неврологических состояниях небольшое снижение уровней 5-MTHF является эпифеноменом, не имеющим патофизиологической значимости. Исходя из опыта авторов, относительно часто слабовыраженный либо умеренно выраженный синдром ЦФН возникает в связи с субоптимальным уровнем фолата в крови на фоне недостатка фолатов в диете (Perez-Dueñas et al, 2011). При диагностике возможного наличия ЦФН следует в первую очередь исключить данное распространенное состояние. Например, недостаточное потребление фолата связано с целым рядом нейропсихиатрических заболеваний (Morris et al, 2002), а фолатная недостаточность и гипергомоцистеинемия часто отмечаются у пожилых пациентов с психиатрическими расстройствами."
Перевод : Тимеев Артём Геннадьевич (переводы фармакологических, биотехнологических текстов с русского на английский и с английского на русский; email: Этот адрес электронной почты защищён от спам-ботов. У вас должен быть включен JavaScript для просмотра.)