Первое описание китайского пациента с ранее неизвестной мутацией гена FOLR1, вызвавшей церебральную фолатную недостаточность
(Скачать перевод в формате PDF)
Цылю Чжан (Ciliu Zhan)1, Сяолу Дэн (Xiaolu Deng)1, Яфэй Вэнь (Yafei Wen)2, Фан Хэ (Fang He)1, Фэй Инь (Fei Yin)1, Цзин Пэн (Jing Peng)1*
Аннотация
Актуальность: Церебральная фолатная недостаточность (ЦФН) – неврологическое заболевание, для которого характерно значительное снижение концентрации 5-метилтетрагидрофолата (5-MTHF) в спинномозговой жидкости (СМЖ). Главные причины ЦФН включают в себя наличие аутоантител к фолатному рецептору (ФР), дефекты гена FOLR1, кодирующего ФР, митохондриальные заболевания и врожденные дефекты фолатного метаболизма.
Описание клинического случая: Мы впервые представляем случай ЦФН у мальчика-китайца, у которого в возрасте 2 лет развились судороги и состояния эпилептического статуса. Магниторезонансная томография (МРТ) выявила признаки развития энцефаломаляции, ламинарного некроза в нескольких долях мозга, и атрофии мозжечка. Полноэкзомное секвенирование (ПЭС) выявило гомозиготный миссенс-вариант c.524G > T (p.C175F) в гене FOLR1. Последующие лабораторные анализы продемонстрировали экстремальное снижение концентрации 5-MTHF в СМЖ пациента, что было сочтено результатом недостаточности церебрального фолатного транспорта. Применение фолината кальция в виде внутривенной терапии и перорально повысило концентрацию 5-MTHF в СМЖ до нормального уровня и привело к ослаблению эпилептических приступов.
Заключение: Представлено первое описание новой вариации гена FOLR1 у китайского мальчика, страдавшего тонико-клоническими судорогами, отставанием в развитии и атаксией. Результаты ПЭС и лабораторных исследований помогли раскрыть причину его симптомов. Ранняя диагностика и назначение корректной терапии улучшили клинический исход.
Ключевые слова: Судороги, FOLR1, 5-MTHF, фолинат кальция
Рукопись получена: 30 июля 2020 года.
Одобрена: 3 ноября 2020 года.
Опубликована онлайн: 26 ноября 2020 года.
Перевод на русский: Тимеев Артем Геннадьевич, Екатеринбург,
Актуальность
Согласно определению, данному в 2004 году Рамакерсом и Блау (Ramaekers and Blau), понятие «церебральная фолатная недостаточность» охватывает группу неврологических синдромов, ассоциированных со сниженной концентрацией 5-метилтетрагидрофолата (5-MTHF), метаболита, необходимого для нормальной работы нервной системы, в спинномозговой жидкости (СМЖ). Косвенные свидетельства указывают на то, что ЦФН может возникать как в виде врожденного метаболического заболевания, так и в качестве приобретенной недостаточности фолата.[2, 3] Самая распространенная причина ЦФН – наличие аутоантител к фолатному рецептору (ФР).[4] ЦФН также может развиваться при наличии дефектов гена FOLR1, при митохондриальных заболеваниях и врожденных нарушениях фолатного метаболизма. Ген FOLR1 (OMIM#613068), расположенный на длинном плече 11-й хромосомы, кодирует фолатный рецептор альфа (FRα).[5] Высокоаффинное связывание FRα c 5-MTHF играет критически важную роль в переносе фолата в мозг.[6, 7] Таким образом, патогенные варианты гена FOLR1 могут приводить к развитию недостаточности 5-MTHF в ЦНС, вызывающей целый ряд нейропсихиатрических симптомов.[5] При мутациях гена FOLR1 церебральная фолатная недостаточность дебютирует обычно в позднем младенческом возрасте,[8] проявляясь в основном следующими симптомами: задержка психического развития, дискинезия, эпилептические приступы, лейкодистрофия и замедление фоновой ЭЭГ-активности. У пациентов младенческого возраста наблюдается крайне низкая концентрация 5-MTHF в спинномозговой жидкости при сохранении нормальной концентрации в периферической нервной системе, что указывает на церебральную недостаточность 5-MTHF как на базовый механизм развития младенческой формы ЦФН.
Описание клинического случая
Пациент
Описываемый пациент китайского происхождения родился доношенным, с массой тела 3.25 кг, что соответствует норме, вагинальные роды протекали без осложнений. Начиная с полутора лет у пациента отмечались отставание в развитии и умственная отсталость. Поднимать голову мальчик начал в возрасте 3 месяцев, сидеть в 6-7 месяцев, ходить в 24 месяца, говорить некоторые слова в 12 месяцев. Начиная с двух лет у мальчика отмечались проблемы со здоровьем.
В возрасте 2 лет у пациента начались тонико-клонические судороги длительностью 20-30 минут, сопровождавшиеся сильной лихорадкой (39°С), изначально диагностированные как фебрильные судороги в больнице по месту жительства. Пациенту были назначены антиконвульсанты. Симптоматика ослабла. Когда пациенту было около четырех лет, у него появились схожие, но более выраженные симптомы. После назначения аналогичной терапии пациент был переведен в другую больницу с целью детальной диагностики. КТ-сканирование не выявило повреждений мозга. Пациенту был поставлен диагноз вирусного менингита и назначена противовирусная терапия. В возрасте 6 лет и 9 месяцев было отмечено ухудшение симптоматики. Пациент переживал приступы рвоты, страдал от головной боли, у него отмечались тонико-клонические судороги и симптоматическая генерализованная эпилепсия с приступами, длящимися до получаса. Пациент был интубирован и получал лечение комбинацией антибиотиков, фенобарбитала и вальпроата натрия.
Несмотря на относительное ослабление симптомов при получении пациентом вальпроата натрия в дозировке 0.2 г три раза в день (26 мг/кг/сутки), причина симптомов оставалась неизвестной. В связи с этим в возрасте 6 лет 11 месяцев, после 4 лет хронических эпизодов эпилептического статуса, пациента направили в больницу Сянъя при Центральном южном университете в провинции Хунань (Китай). Тщательное неврологическое обследование выявило клинически выраженную общую задержку развития, нечленораздельную речь, гиперактивность, гиперсаливацию, затруднения с контролем поведения, нетвердую походку и мышечную гипотонию. Окружность головы составляла 46 см (ниже 3-й процентили). Для выявления причин заболевания были назначены подробные лабораторные и генетические исследования.
Было осуществлено диффузно-взвешенное МРТ-сканирование (DWI-сканирование). Сканирование выявило признаки энцефаломаляции и ламинарного некроза в височно-теменной доле слева, в гиппокампе и во фронтальной доле билатерально с диффузными изменениями белого вещества мозга (рисунок 1a). Также была выявлена атрофия мозжечка (рисунок 1b). Магнитно-резонансная ангиография (МРА) черепно-мозговой области не выявила отклонений. Сбор и анализ клинических данных о семье пациента происходил с одобрения Этического комитета больницы Сянъя при Центральном южном университете. Информированное согласие получали у родителей пробанда.
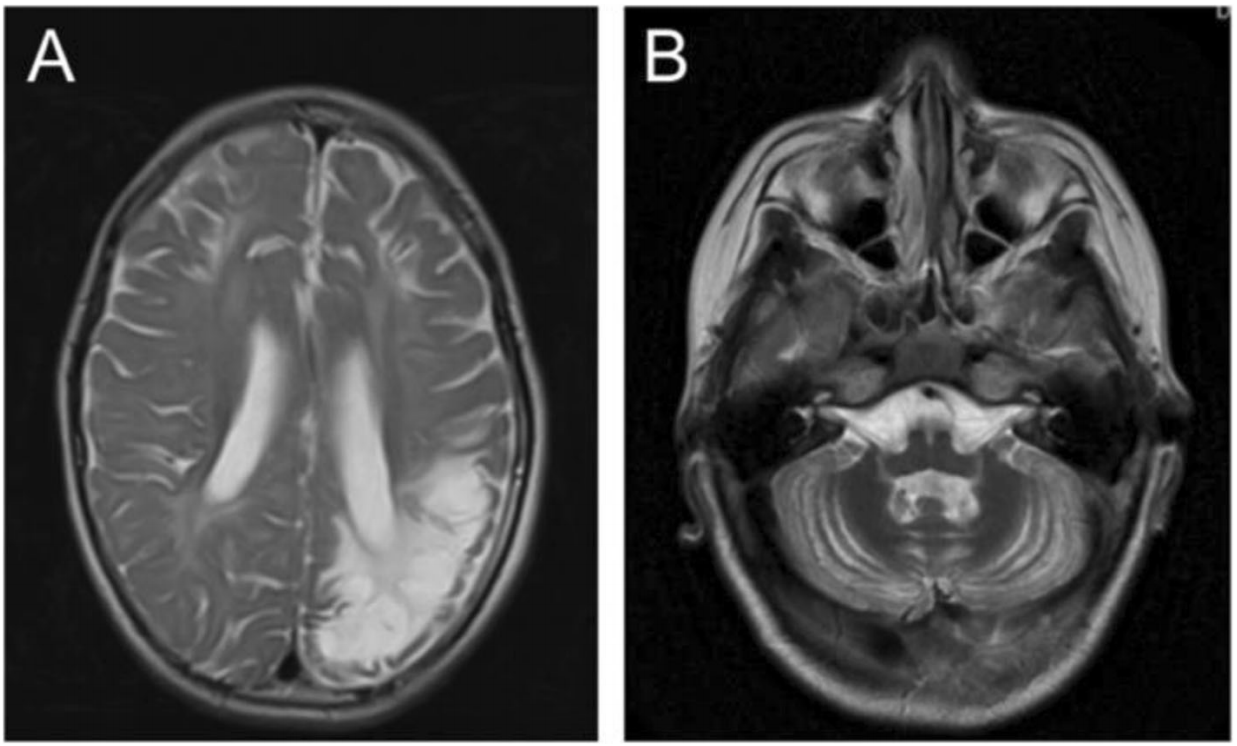
Рис. 1. Контрастная МРТ мозга мальчика в возрасте 6 лет 11 месяцев. a) МРТ-снимок демонстрирует энцефаломаляцию и ламинарный некроз с диффузными изменениями белого вещества. b) На МРТ-снимке видна дистрофия мозжечка.
Генетический анализ
В качестве беспристрастного метода поиска потенциальных патогенных вариантов применили полноэкзомное секвенирование (ПЭС). Геномную ДНК извлекали из образцов периферической крови пробанда и его родителей в соответствии с ранее описанной процедурой.[8] После этого осуществляли ПЭС-скрининг для выявления потенциальных мутаций в геномной ДНК. Сиквенсные варианты проверяли с помощью популяционных баз данных gnomAD (http://gnomad.broadinstitute.org/) и оценивали с помощью различных биоинформационных программ. Патогенность вариантов оценивали в соответствии с руководством Американского колледжа медицинской генетики (ACMG).[9] Производили дополнительную проверку обнаруженного варианта, производя секвенирование по Сенгеру.
Был выявлен новый вариант c.524G > T (p.C175F) в гене фолатного рецептора альфа (FOLR1). Секвенирование по Сенгеру подтвердило гомозиготность варианта в геноме пациента, при этом было установлено, что его родители являются гетерозиготными носителями мутации (рисунок 2a). Вариант не обнаруживался в популяционной базе данных gnomAD. Анализ с помощью нескольких биоинформационных инструментов (Балл Polyphen2 = 0.995, вероятно патогенный; Балл Mutation Taster = 0.998, патогенный; Балл SIFT = 0, патогенный) указывает на патогенный характер обнаруженного варианта.
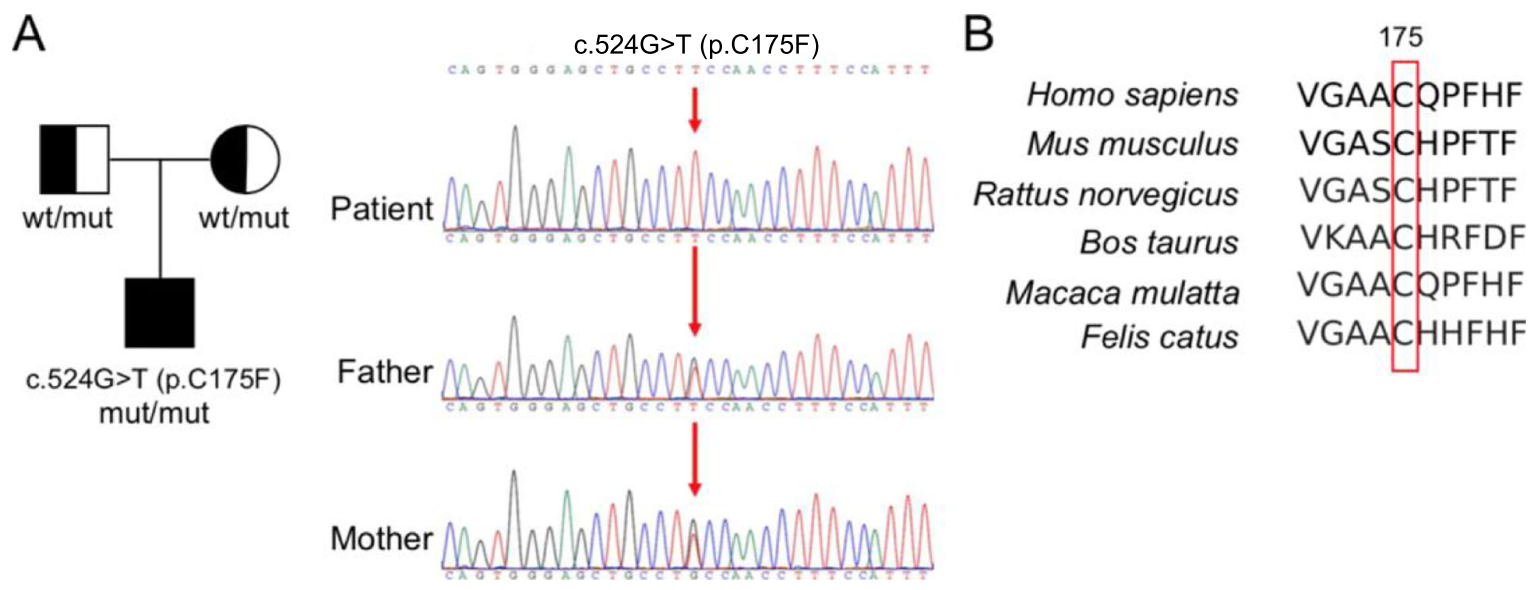
Рис. 2. Полноэкзомное секвенирование (ПЭС) и секвенирование по Сенгеру выявили миссенс-вариант c.524G > T (p.C175F) в гене FOLR1. a) Пробанд является гомозиготным носителем варианта, в то время как его родители являются гетерозиготными носителями. b) Эволюционная консервативность цистеинового остатка в позиции 175 (красная рамка) гена FOLR1 на примере различных видов. wt - wildtype, вариант «дикого типа».
Последующая диагностика
По данным стандартного лабораторного анализа образцов крови, концентрация общего фолата в крови пациента составляла 2.20 мкг/л, что не превышает нижнюю границу нормы (3 мкг/л), а концентрация внутриклеточного фолата составляла 82.66 мкг/л, что также ниже нижней границы нормы (93 мкг/л).
Дальнейшее исследование показало, что в спинномозговой жидкости пациента почти не обнаруживается 5-MTHF. Средняя концентрация 5-MTHF составила 1.38 нмоль/л, что намного ниже нормального диапазона (60 – 210 нмоль/л) для детей в возрасте от 6 до 15 лет. Эти результаты указывали на наличие церебральной фолатной недостаточности. Взятые в совокупности, результаты лабораторных анализов и ПЭС указывали на мутацию в гене FOLR1 и связанную с ней недостаточность 5-MTHF в СМЖ пациента как на основную причину его симптомов.
Терапия
На основании полученных результатов генетического и лабораторного тестирования мы выбрали следующую тактику лечения: кальция фолинат внутривенно (в/в) в дозировке 2 мг/кг/сутки на протяжении 1-й недели с доведением дозировки до 6 мг/кг/сутки с помощью орального приема, с последующим плавным переходом на дозу 11 мг/кг/сутки перорально. Во время терапии фолинатом кальция осуществлялся тщательный мониторинг концентрации 5-MTHF в СМЖ. Дозировку противоэпилептических препаратов (ПЭП) постепенно снижали до полной ремиссии эпилепсии. После 2 месяцев терапии концентрация 5-MTHF в СМЖ выросла до 36.24 нмоль/л, а еще через 6 месяцев – до 78.76 нмоль/л, что укладывается в нормальный диапазон концентраций, 60-210 нмоль/л, установленный для детей в возрасте от 6 до 15 лет. Параллельно отмечалось значительное ослабление таких симптомов, как нетвердость походки, неспособность уверенно держать предметы, эпилептические приступы и т.д. Более того, после назначения терапии судороги более не сопровождались лихорадкой.
Выводы и обсуждение
В настоящем отчете мы описали новую мутацию гена FOLR1, вызвавшую церебральную фолатную недостаточность у китайского пациента (рисунок 1a). Вплоть до выяснения причины симптомов юного пациента переводили из одной больницы в другую, и у него наблюдались частые рецидивы. Благодаря обнаружению мутации гена FOLR1, связанной с клиническими проявлениями заболевания, удалось оперативно начать терапию церебральной фолатной недостаточности у пациента.
Продукт гена FOLR1 представляет собой гликозилфосфатидилинозитол-заякоренный (ГФИ-заякоренный) мембранный белок, регулирующий процесс переноса фолата в клетки.[6] Кристаллическая структура человеческого белка FOLR1 в комплексе с фолиевой кислотой была изучена группой Chen et al. при разрешении в 2.8 Å.[7] Другая группа ученых определила дискретные структурные конформации белка при разных значениях pH.[10] Отсутствуют непосредственные свидетельства того, что новый вариант c.524G > T (p.C175F) влияет на связывание лиганда. Вместе с тем в геноме различных видов сохраняется вариант C175, а клинические данные, полученные в ходе настоящего исследования, демонстрируют важность данного аминокислотного остатка. Возможно, мутация в этой позиции каким-то образом нарушает работу белка. Будущие опыты должны пролить свет на то, как этот вариант влияет на функционирование FOLR1 и на фолат-связывающую способность белка.
В генетическом локусе FOLR1 описаны патогенные варианты, ассоциированные с развитием неврологических заболеваний.[2, 5, 8, 10, 11] Гомозиготные мутации либо компаунд-гетерозиготные мутации вызывают заболевания, наследуемые по аутосомно-рецессивному типу. Также выявлена гомозиготная внутрирамочная дупликация размером 18 пар оснований, ассоциированная с нейродегенеративным заболеванием.[5] Данные указывают на то, что у всех носителей мутаций гена FOLR1 обнаруживается значительное снижение концентрации 5-MTHF в СМЖ (≤ 5 нмоль/л), что приводит к выявлению множества очагов повреждения в головном мозге при сканировании мозга. Осуществив обзор публикаций, мы составили сводку 15 вариантов, описанных у 23 пациентов – она представлена графически на рисунке 3.[2, 5, 8, 10, 12–17] Мы добавили обнаруженную новую гомозиготную вариацию p.C175F гена FOLR1 в генотипный спектр. Сравнение белковых последовательностей человеческого FOLR1 с последовательностями аналогичных белков других организмов показывает, что остаток C175 «законсервирован», он обнаруживается у различных видов, следовательно, данный сайт белка крайне необходим для его нормальной работы и мутация в этой позиции может нарушать связывание и транслокацию 5-MTHF. Для того чтобы скорректировать вызванное мутацией p.C175F снижение концентрации 5-MTHF, мы применили фолинат кальция, рацемически стабильную форму фолата, в форме внутривенного раствора и в оральной форме.[18] Терапевтическая схема эффективно ослабила симптомы заболевания. Учитывая возможную нейротоксичность избыточных доз фолиевой кислоты,[19] важно отличать фолиниевую кислоту от фолиевой, помня и про то, что последняя может ухудшить симптомы, уменьшая пропорцию активных фолатных рецепторов за счет сильного связывания с ними. В этой связи мы осуществляли тщательный мониторинг изменений концентрации 5-MTHF в СМЖ, чтобы не допустить каких-либо побочных эффектов от терапии.
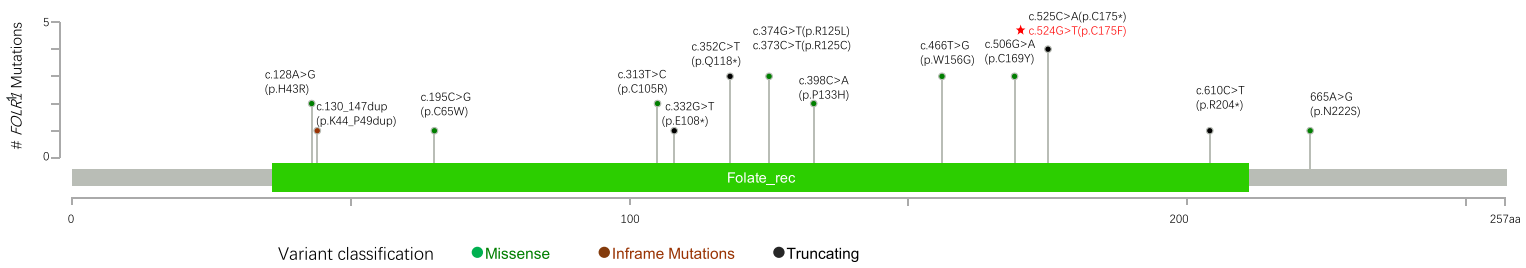
Рис. 3. График типа «лоллипоп», демонстрирующий мутации гена FOLR1, описанные в научной литературе. Красной звездочкой помечен вариант, выявленный в настоящем исследовании. Зеленые точки: миссенс-мутации; красные точки: мутации внутри рамки считывания; синие точки: усекающие мутации. Зеленая полоса: Фолат-связывающий домен семейства фолатных рецепторов. Примечание: сплайс-мутация g.3576 T > G (упоминается в работе [8]) не отображена на графике, поскольку при реаннотации генома было установлено, что она не затрагивает данный ген.
Итак, для выбора эффективной терапии у пациента с частыми рецидивами эпилепсии исключительно важно понимать генетические предпосылки заболевания. Такое понимание может быть крайне полезно для юных пациентов, поскольку раннее начало эффективной терапии способствует лучшему восстановлению.
Благодарности
Мы благодарим пациента и его семью за их сотрудничество и вклад в исследование. Благодарим доктора Сяодун Вана (Xiaodong Wang) из компании Cipher Gene, Inc. за помощь в редактировании рукописи.
Сокращения
ЦФН
Церебральная фолатная недостаточность
5-MTHF
5-метилтетрагидрофолиевая кислота
СМЖ
Спинномозговая жидкость, ликвор
ФР
Фолатный рецептор
МРТ
Магниторезонансная томография
ПЭС
Полноэкзомное секвенирование
DWI-сканирование
Диффузно-взвешенное МРТ-сканирование
ACMG
Американский колледж медицинской генетики
ПЭП
Противоэпилептические препараты
Вклад авторов
Автор CZ (Цылю Чжан) была лечащим врачом пациента. Цылю Чжан анализировала данные, полученные при секвенировании методом Сенгера, продумала концепцию публикации, составила первоначальную рукопись и занималась редактурой статьи. Авторы XD и YW помогали в сборе клинической информации, обеспечивали дальнейшее наблюдение за пациентом, интерпретировали данные. Автор FH анализировал результаты МРТ-сканирования пациентов. Автор FY предоставлял консультации по данному клиническому случаю, высказывал предложения по терапии и занимался редактурой публикации. Автор JP отвечал за анализ данных, полученных при секвенировании методом Сенгера, написание рукописи, выполнение критической редактуры рукописи. Все авторы ознакомились с рукописью и одобрили ее.
Финансирование
Выполнение работы было спонсировано Хунаньским провинциальным отделением Фонда естественных наук Китая (грант № 2020JJ5946) и Национальным фондом естественных наук Китая (гранты № 81801297; № 81771408). Полноэкзомное секвенирование было проведено за счет полученного финансирования.
Доступность данных и материалов.
Автор, отвечающий за корреспонденцию, предоставляет данные, связанные с настоящим исследованием, в ответ на обоснованный запрос.
Данные были загружены в базу данных NCBI SRA и свободно доступны по следующей ссылке: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA672777
Соответствие этическим нормам и получение согласия на участие
Все процедуры соответствовали этическим стандартам в отношении экспериментов с участием людей, установленным ответственным Комитетом по этике при больнице Сянъя, Центральный южный университет (округ Чанша, Китай), а также положениям Хельсинской декларации. Для выполнения настоящей работы у каждого участника получали информированное согласие. В отношении участников, не достигших 16 лет, информированное согласие предоставляли родители.
Согласие на публикацию
Для публикации настоящего клинического случая было получено информированное согласие в письменной форме от родителей пациента. Копию подписанной формы информированного согласия можно получить для ознакомления у редактора данного журнала.
Конфликт интересов
Авторы заявляют об отсутствии конфликтов интересов.
Комментарий издателя
Издательство Springer Nature придерживается нейтральности в отношении утверждений о границах юрисдикций в публикуемых картах, а также в отношении утверждений о принадлежности авторов к институтам.
Список литературы
- Ramaekers VT, Blau N. Cerebral folate deficiency. Dev Med Child Neurol. 2004;46:843–851. doi: 10.1111/j.1469-8749.2004.tb00451.x. [PubMed] [CrossRef] [Google Scholar]
- Cario H, Bode H, Debatin K-M, Opladen T, Schwarz K. Congenital null mutations of the FOLR1 gene: a progressive neurologic disease and its treatment. Neurology. 2009;73:2127–2129. doi: 10.1212/WNL.0b013e3181c679df. [PubMed] [CrossRef] [Google Scholar]
- Desai A, Sequeira JM, Quadros EV. The metabolic basis for developmental disorders due to defective folate transport. Biochimie. 2016;126:31–42. doi: 10.1016/j.biochi.2016.02.012. [PubMed] [CrossRef] [Google Scholar]
- Frye RE, Slattery JC, Quadros EV. Folate metabolism abnormalities in autism: potential biomarkers. Biomark Med. 2017;11:687–699. doi: 10.2217/bmm-2017-0109. [PubMed] [CrossRef] [Google Scholar]
- Steinfeld R, Grapp M, Kraetzner R, Dreha-Kulaczewski S, Helms G, Dechent P, et al. Folate receptor alpha defect causes cerebral Folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am J Hum Genet. 2009;85:354–363. doi: 10.1016/j.ajhg.2009.08.005. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Della-Longa S, Arcovito A. Structural and functional insights on folate receptor α (FRα) by homology modeling, ligand docking and molecular dynamics. J Mol Graph Model. 2013;44:197–207. doi: 10.1016/j.jmgm.2013.05.012. [PubMed] [CrossRef] [Google Scholar]
- Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, et al. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013;500:486–489. doi: 10.1038/nature12327. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Grapp M, Just IA, Linnankivi T, Wolf P, Lücke T, Häusler M, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135:2022–2031. doi: 10.1093/brain/aws122. [PubMed] [CrossRef] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Al-Baradie RS, Chaudhary MW. Diagnosis and management of cerebral folate deficiency. A form of folinic acid-responsive seizures. Neurosciences (Riyadh) 2014;19(4):312–316. [PMC free article] [PubMed] [Google Scholar]
- Dill P, Schneider J, Weber P, Trachsel D, Tekin M, Jakobs C, et al. Pyridoxal phosphate-responsive seizures in a patient with cerebral folate deficiency (CFD) and congenital deafness with labyrinthine aplasia, microtia and microdontia (LAMM) Mol Genet Metab. 2011;104:362–368. doi: 10.1016/j.ymgme.2011.05.019. [PubMed] [CrossRef] [Google Scholar]
- Kobayashi Y, Tohyama J, Akiyama T, Magara S, Kawashima H, Akasaka N, et al. Severe leukoencephalopathy with cortical involvement and peripheral neuropathy due to FOLR1 deficiency. Brain and Development. 2017;39:266–270. doi: 10.1016/j.braindev.2016.09.011. [PubMed] [CrossRef] [Google Scholar]
- Pérez-Dueñas B, Toma C, Ormazábal A, Muchart J, Sanmartí F, Bombau G, et al. Progressive ataxia and myoclonic epilepsy in a patient with a homozygous mutation in the FOLR1 gene. J Inherit Metab Dis. 2010;33:795–802. doi: 10.1007/s10545-010-9196-1. [PubMed] [CrossRef] [Google Scholar]
- Delmelle F, Thöny B, Clapuyt P, Blau N, Nassogne M-C. Neurological improvement following intravenous high-dose folinic acid for cerebral folate transporter deficiency caused by FOLR-1 mutation. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2016;20:709–713. doi: 10.1016/j.ejpn.2016.05.021. [PubMed] [CrossRef] [Google Scholar]
- Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N, Shimozawa N, Takamura A, Doi H, Tomita-Katsumoto A, Nishiyama K, Tsurusaki Y, Nakashima M, Miyake N, Eto Y, Tanaka F, Matsumoto N, Saitsu H. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics. 2013;14(3–4):225–232. doi: 10.1007/s10048-013-0375-8. [PubMed] [CrossRef] [Google Scholar]
- Ferreira P, Luco SM, Sawyer SL, Davila J, Boycott KM, Dyment DA. Late diagnosis of cerebral folate deficiency: fewer seizures with folinic acid in adult siblings. Neurol Genet. 2015;2(1):e38. doi: 10.1212/NXG.0000000000000038. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Toelle SP, Wille D, Schmitt B, Scheer I, Thöny B, Plecko B. Sensory stimulus-sensitive drop attacks and basal ganglia calcification: new findings in a patient with FOLR1 deficiency. Epileptic Disord Int Epilepsy J Videotape. 2014;16:88–92. doi: 10.1684/epd.2014.0629. [PubMed] [CrossRef] [Google Scholar]
- Ormazabal A, Artuch R, Vilaseca MA, Aracil A, Pineda M. Cerebrospinal fluid concentrations of folate, biogenic amines and pterins in Rett syndrome: treatment with folinic acid. Neuropediatrics. 2005;36:380–385. doi: 10.1055/s-2005-873078. [PubMed] [CrossRef] [Google Scholar]
- Shorvon SD. The etiologic classification of epilepsy. Epilepsia. 2011;52:1052–1057. doi: 10.1111/j.1528-1167.2011.03041.x. [PubMed] [CrossRef] [Google Scholar]
© Автор(ы). 2020
Открытый доступ. Настоящая статья опубликована по лицензии Creative Commons Attribution 4.0 International License, позволяющей использовать, делиться, адаптировать, распространять и воспроизводить материал с использованием любого носителя и в любом формате при условии включения надлежащей ссылки на исходный материал и указания авторства исходного материала, предоставления ссылки на текст используемой лицензии Creative Commons, и обозначения факта внесения изменений в материал. Лицензия Creative Commons, по которой опубликована статья, распространяется на все изображения и иные материалы данной статьи, созданные третьими сторонами, за исключением случаев, когда обратное указано в пояснении к конкретному материалу. Если на материал не распространяется лицензия Creative Commons, распространяющаяся на статью, и планируемое вами использование материала противоречит существующим нормативным правовым актам либо выходит за рамки допустимого использования, вам следует обратиться за разрешением напрямую к правообладателю. Ознакомиться с текстом лицензии можно по следующему адресу: http://creativecommons.org/licenses/by/4.0/. На все данные, приведенные в настоящей статье, распространяется заявление об отказе от авторских прав (Creative Commons Public Domain Dedication, доступно по адресу http://creativecommons.org/publicdomain/zero/1.0/), если иное не указано в пояснении к конкретному набору данных.
* Адрес для переписки:
1Xiangya Hospital Central South University, 87 Xiangya Road, Changsha, Hunan 410008, P.R. China
2XiangYa School of Medicine of Central South University, 172 Tongzipo Road, Changsha, Hunan 410013, P.R. China.