Церебральная фолатная недостаточность у взрослых пациентов: гетерогенное состояние, допускающее возможность терапии
(Cerebral folate deficiency in adults: A heterogeneous potentially treatable condition; M. Masingue et al., Journal of the Neurological Sciences, 2019)
Аннотация
Цель: Описать фенотип и реакцию на назначение фолиниевой кислоты при взрослой форме церебральной фолатной недостаточности (ЦФН) – заболевания, диагностируемого по сниженному содержанию 5-метилтетрагидрофолата (5-MTHF) в спинномозговой жидкости (ликворе), и развивающегося по причине наследуемого дефекта фолатного метаболизма (НДФМ) либо вследствие ряда неврологических заболеваний.
Методы: Мы провели ретроспективное исследование 224 пациентов взрослого возраста с неврологическими симптомами, в отношении которых имелись данные о концентрации 5MTHF в ликворе. В ходе исследования осуществлялся сбор неврологических данных и снимков мозга пациентов.
Результаты: ЦФН (концентрация 5MTHF в ликворе < 41 нмоль/л) была выявлена у 69 пациентов, из них 25 страдали тяжелой ЦФН (тЦФН; ≤25 нмоль/л), причем в последней группе дебют неврологических симптомов во взрослом возрасте наблюдался у 41% пациентов. Исходное неврологическое заболевание удалось установить у 56% пациентов с тЦФН – как правило, это были митохондриальные заболевания, печеночная энцефалопатия и первичные кальцификации структур головного мозга (в отсутствие подтвержденной наследственной дисфункции фолатного метаболизма). Остальные случаи были сочтены «случаями с нераспознанной причиной». Обследование пациентов с тЦФН наиболее часто выявляло наличие пирамидного синдрома (75%), двигательных расстройств (56%), мозжечкового синдрома (50%) и умственной отсталости (46%). При МРТ-исследовании в 23% случаев признаки патологии отсутствовали, а при их наличии основными отклонениями были нарушения целостности белого вещества (НЦБВ; 32%) и наличие кальцификаций (12%). Не было выявлено явных отличий клинико-радиологического фенотипа пациентов с тЦФН от такового у пациентов без ЦФН в плане частоты патологических проявлений. Вместе с тем у первых наблюдалась более сложная неврологическая картина с повышенным количеством комбинированных неврологических симптомов (4.7 ± 1.6 против 3.4 ± 1.7, p = .01). При исследовании с помощью магниторезонансной спектроскопии (МРС) у пациентов с тЦФН (n = 7) было выявлено более низкое отношение холин/креатин (Cho/Cr), чем у пациентов без ЦФН (n = 73) – этому различию была свойственна хорошая чувствительность (71%) и прекрасная специфичность (92%). Назначение фолиниевой кислоты двадцати одному пациенту с ЦФН привело к устойчивому улучшению симптоматики у девяти пациентов, восемь из которых страдали тЦФН (улучшение продемонстрировали 50% пациентов с тЦФН). У двух пациентов, у которых по неизвестной причине наблюдались экстремально низкие концентрации 5MTHF в ликворе вкупе с нарушениями целостности белого вещества (МРТ) и сниженными соотношениями Cho/Cr, применение фолиниевой кислоты привело к драматическому улучшению клинической симптоматики и результатов радиологических исследований.
Выводы: при наличии у пациента митохондриального заболевания, первичной кальцификации структур мозга, либо сложного неврологического расстройства невыясненной этиологии, особенно вкупе с нарушениями целостности белого вещества мозга, следует рассмотреть возможность измерения концентрации 5MTHF в ликворе, поскольку при тяжелой церебральной фолатной недостаточности восполнение уровней фолата с помощью фолиниевой кислоты часто оказывается эффективным.
Сокращения: 5MTHF – 5-метилтетрагидрофолат; ЦФН – церебральная фолатная недостаточность; тЦФН – тяжелая церебральная фолатная недостаточность; Cho/Cr – отношение холин/креатинин; ликвор – спинномозговая жидкость; FOLR1 – фолатный рецептор 1; FRα – фолатный рецептор-альфа; ПЭ – печеночная энцефалопатия; УО – умственная отсталость; НДФМ – наследственные дефекты фолатного метаболизма; СКС – синдром Кернса-Сейра; МРТ – магниторезонансная томография; МРС – магниторезонансная спектроскопия; MTHFR – метилтетрагидрофолатредуктаза; ПКМ – первичная кальцификация структур мозга; ФГК1 – фосфоглицераткиназа 1; ПОЛГ – ДНК-полимераза гамма (каталитическая субъединица полимеразы митохондриальной ДНК); СПГ 11 – спастическая параплегия 11, *Автор, осуществляющий корреспонденцию.
1. Введение.
Церебральная фолатная недостаточность (ЦФН) – состояние, характеризуемое сниженной концентрацией 5-метилтетрагидрофолата (5MTHF) в спинномозговой жидкости (ликворе) – диагностируется у детей, страдающих различными неврологическими расстройствами, и может сопровождаться либо не сопровождаться периферическим дефицитом фолата. [1–3]. 5MTHF участвует в синтезе ДНК, аминокислот, белков и нейромедиаторов и выступает донором метильных групп в процессе реметилирования гомоцистеина [4].
После всасывания стенками кишечника фолат восстанавливается до 7,8-дигидрофолата (DHF), затем до 5,6,7,8-тетрагидрофолата (THF) в реакции, катализируемой ферментом дигидрофолатредуктазой (DHFR). После этого серин действует в роли донора метильной группы в реакции, катализируемой ферментом серин-гидроксиметилтрансферазой (SHMT), преобразующей THF в 5,10-метилен-THF, а также в реакции, образующей глицин; фермент метилентетрагидрофолатредуктаза (MTHFR) преобразует 5,10-метилен-THF в 5MTHF – активную и наиболее стабильную циркулирующую форму фолата [5].
Перенос 5MTHF из кровообращения в ликвор происходит в основном с помощью фолатного рецептора альфа (FRα) [5–7]. Благодаря процессу FRα-опосредованного эндоцитоза, расходующему АТФ, концентрация 5MTHF в ликворе может втрое превышать концентрацию 5MTHF в крови [2]. PCFT (протон-сопряженный транспортер фолата) и RFC (восстановленный переносчик фолата) – два других транспортера фолатов, которые также присутствуют в эпителиальных клетках сосудистого сплетения – отличаются более низким сродством к фолату по сравнением с FRα, и участвуют в основном в кишечной абсорбции фолата [6,7].
Наследуемые дефекты фолатного метаболизма (НДФМ) могут приводить к развитию ЦФН [5]. Врожденная мальабсорбция фолатов, связанная с дефектами гена PCFT, приводит к развитию мегалобластной анемии с иммунной недостаточностью, сопровождающейся судорогами и когнитивными нарушениями [8]. Недостаточность дигидрофолатредуктазы (DHFR) проявляется развитием мегалобластной анемии и эпилепсии в младенческом возрасте [9]. Недостаточность фермента MTHFR ассоциирована со снижением когнитивных функций, спастическим парапарезом, периферической нейропатией, эпилепсией и лейкоэнцефалопатией [10]. При этих трех заболеваниях ЦФН обычно сопровождается сниженной концентрацией фолата в крови. Мутации, связанные с потерей функции гена FOLR1 (кодирующего белок FRα), приводят к развитию в младенческом возрасте прогрессирующего мозжечкового синдрома с двигательными нарушениями, сопровождающегося эпилептическими приступами, регрессией психологического развития, и признаками гипомиелинизации и атрофии головного мозга на МРТ-снимках. Уровень фолата в образцах сыворотки крови при этом остается в рамках нормы [11-14]. Помимо этих НДФМ, случаи развития синдрома ЦФН, как правило, не сопровождающиеся периферической недостаточностью фолата, были отмечены у детей, страдающих генетически обусловленными заболеваниями, особенно митохондриальными заболеваниями (в том числе синдромом Кернса-Сейра[5,15,16]), синдромом Ретта, синдромом Айкарди–Гутьерес [5]. Также были отмечены идиопатические случаи развития синдрома, проявляющиеся задержкой психомоторного развития, тугоухостью, мозжечковой атаксией, пирамидным синдромом, поведенческими отклонениями или эпилепсией [2,19]. В некоторых работах делаются предположения о возможности аутоиммунного развития ЦФН ввиду обнаружения у пациентов антител к FRα, однако эта гипотеза вызывает споры [2,5,18,20–22]. У многих детей с неврологической симптоматикой конкретные причины синдрома ЦФН остаются невыясненными.
Терапия ЦФН заключается в пероральном приеме больших доз фолиниевой кислоты. Согласно исследованиям, препарат позволяет нормализовать концентрацию 5MTHF в ликворе и в некоторых случаях приводит к улучшению клинической симптоматики и результатов сканирования мозга [13,15,23,24].
К настоящему времени синдром ЦФН описан только у детей, за исключением одного случая, в котором было описано развитие синдрома в возрасте 58 лет в виде таких симптомов, как прогрессирующая потеря памяти и миоклония, при этом назначение фолиниевой кислоты привело к улучшению состояния пациента [25].
Задавшись целью исследовать фенотипический спектр ЦФН у взрослых пациентов, мы осуществили ретроспективный сбор клинических и радиологических данных группы пациентов с неврологическими симптомами, в отношении которых были известны уровни концентрации 5MTHF в ликворе.
2. Материалы и методы
Нами был осуществлен ретроспективный сбор клинической и радиологической информации о пациентах, проходивших обследование в больнице Питье-Сальпетриер (Париж, Франция), включавшее измерение концентрации 5MTHF в ликворе в период с 2008 по 2015 годы. Концентрация 5MTHF в ликворе у данных пациентов измерялась в ходе рутинного обследования по поводу неврологического синдрома неясной этиологии, а в некоторых случаях – в ходе последующего наблюдения по поводу ЦФН. Все анализы концентрации, за исключением трех, были выполнены в Отделении биохимии Больницы им. Роберта Дебре (JFB). Два из этих трех анализов были выполнены в Больнице им. Труссо (FM), а один – в Университетской детской больнице Цюриха (Ненад Блау / Nenad Blau). Концентрацию 5MTHF в ликворе определяли методом высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией в режиме регистрации положительно заряженных ионов. Наблюдается обратная корреляция между возрастом и верхней границей нормальной концентрации 5MTHF: ликворная концентрация 5MTHF в норме у детей выше, чем у взрослых [26]. Экстраполируя степень снижения ликворной концентрации 5MTHF, наблюдаемую в детской популяции, мы установили пороговое значение 5MTHF для диагностики ЦФН у взрослых на уровне 41 нмоль/л. Это значение уже было использовано в ряде научных публикаций [25–28]. Мы произвольно установили порог, равный 25 нмоль/л, для определения подгруппы пациентов с тяжелой ЦФН (с концентрацией 5MTHF в ликворе < 25 нмоль/л) – эти пациенты были нашим основным объектом исследования – поскольку, по нашему представлению, ЦФН умеренной степени тяжести, с уровнем фолата в диапазоне 25-41 нмоль/л, не приводит к клинически заметным проявлениям. Этот произвольно выбранный уровень оказался подходящим, поскольку почти все пациенты с ЦФН, достигшие улучшения на заместительной фолатной терапии, страдали от тяжелой формы ЦФН (результаты приведены ниже), а пациенты с ЦФН умеренной степени тяжести не продемонстрировали улучшений. Помимо 5MTHF, была осуществлена оценка ликворной концентрации 5-гидроксииндолуксусной кислоты (5HIAA, метаболит серотонина) и гомованилиновой кислоты (HVA, метаболит дофамина). Не было отмечено корреляции между концентрацией 5MTHF и концентрацией HVA, а между концентрациями 5MTHF и HIAA была отмечена слабая обратная корреляция (r = 0.145, p = .036). Сто пациентов подверглись магниторезонансной спектроскопии (МРС) для анализа соотношений холина/креатина (Cho/Cr), N-ацетиласпартата(NAA)/креатина, и холина/NAA в белом веществе мозга (овальный либо полуовальный центр) с длинным временем эхо (TR = 1500 мс, TE = 135 мс). Для определения нормальных соотношений брались результаты 12 контрольных лиц без неврологических заболеваний, подобранных по возрасту. В частности, для отношения Cho/Cr был выбран нормальный диапазон ≥0.90.
В ходе статистического анализа с помощью пакета IBM SPSS Statistics 24.0.0 производилась, по мере необходимости, оценка критерия хи-квадрат, критерия Фишера, и критерия Манна-Уитни. Корреляция оценивалась с помощью коэффициента корреляции Спирмена.
3. Результаты
Всего скринингу на ЦФН подверглось 224 взрослых пациента. Было выявлено 69 пациентов с ЦФН, в том числе 25 пациентов с тяжелой ЦФН (тЦФН; Таблица 1). Средний возраст на момент начала заболевания составил 24 года (± 21, 0–65). Среди пациентов с тЦФН болезнь чаще начиналась в детском возрасте (< 15 лет; 13/22, 59%), чем среди пациентов с нетяжелой формой ЦФН (15/40, 37.5%). Данные о концентрации фолата в крови были доступны в отношении 38/69 пациентов в целом, и в отношении 15/25 пациентов с тЦФН в частности. Нормальный уровень был отмечен у 36 пациентов, повышенный – у одного пациента, пониженный – у одного пациента. Среди 31 пациента с ЦФН, для которых отсутствовали данные о концентрации фолата в образцах сыворотки крови, у 22 не было отмечено системных проявлений либо биологически выявляемых отклонений (анемии и/или повышенных уровней гомоцистеина), на основании которых можно было предположить наличие фолатной недостаточности, у одного была выявлена макроцитарная анемия, а в отношении остальных (n=8) отсутствовали результаты соответствующих анализов. Среди пациентов, относительно которых была доступна информация о получаемой ими терапии, не было отмечено случаев получения препаратов, вмешивающихся в метаболизм фолата, таких как метотрексат, пириметамин или триметоприм.
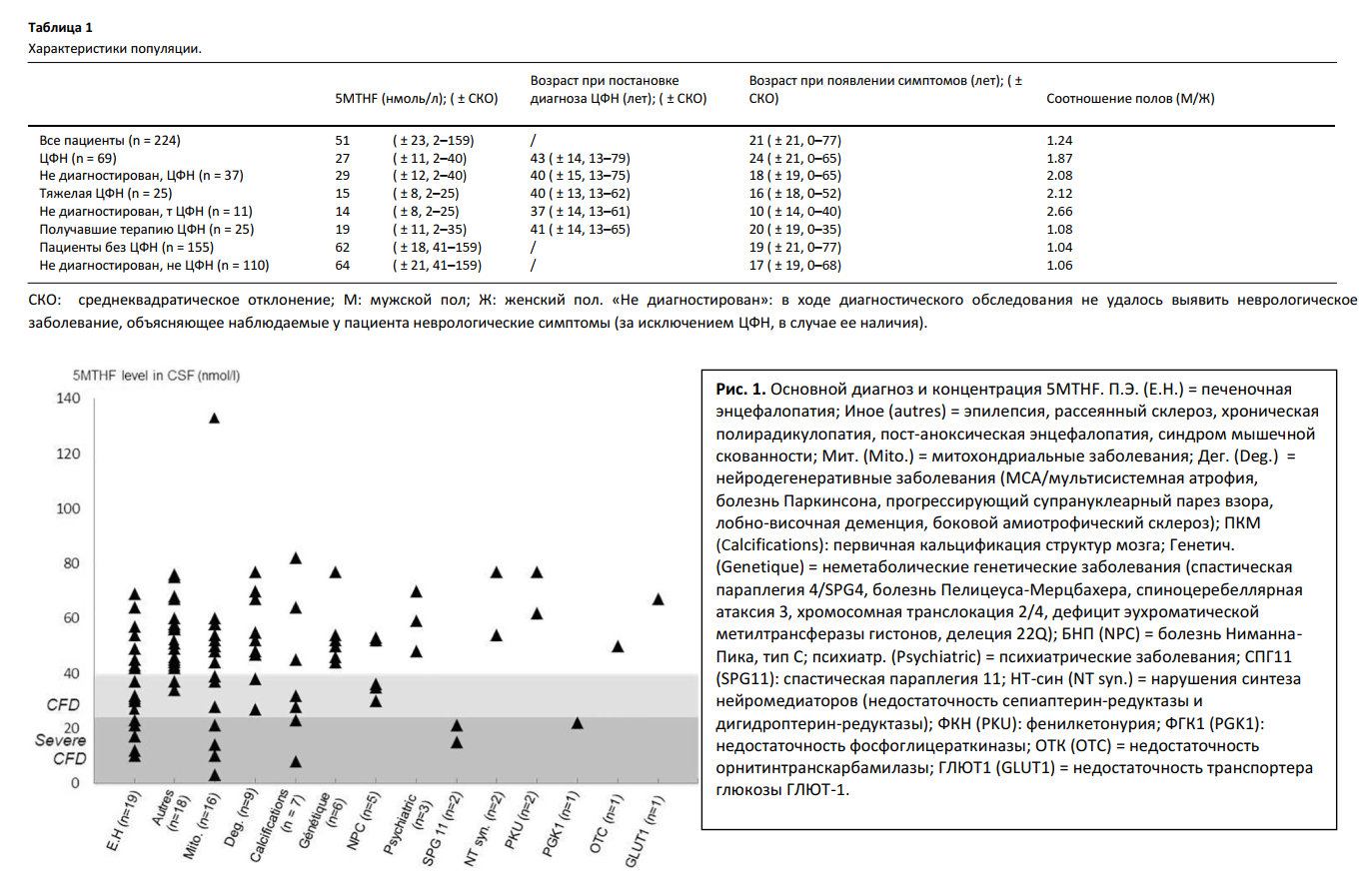
3.1. Ассоциированные заболевания
В группе пациентов с тЦФН (n = 25), 14 (56%) пациентов имели установленный диагноз (Рис. 1). Митохондриальные заболевания были выявлены у четырех пациентов: у одного – мутация в гене ДНК-полимеразы гамма (POLG), у одного – синдром Кернса-Сейра, и у двух – убедительные признаки митохондриального заболевания (включая результаты биопсии мышц) в отсутствие доказанных генетических нарушений. Другие диагнозы включали первичную кальцификацию структур мозга (ПКМ, n = 2) без установленных генетических вариаций, мутации гена SPG11 (n = 2), мутации гена PGK1 (n = 1), и печеночную энцефалопатию (ПЭ; n = 5). Скрининг на наличие мутаций гена FOLR1 дал отрицательный результат у десяти пациентов с ЦФН (в том числе шести пациентов с тЦФН). Анализ на антитела к FRα был проведен девяти пациентам, и оказался положителен у семи из них. Вместе с тем у двух пациентов, которым анализ был проведен повторно, наличие антител подтверждено не было.
Примечание: два пациента с мутацией гена SPG11 – братья.
3.2. Клинический фенотип
У пациентов с тЦФН отмечались в основном пирамидные симптомы (n = 18, 75%), гиперкинезы (n = 14, 56%), мозжечковый синдром (n = 12, 50%), умственная отсталость (УО; n = 11, 46%), снижение когнитивных функций (n = 9, 38%), периферическая нейропатия (n = 6, 25%), нарушение слуха (n = 6, 25%) и эпилепсия (n = 6, 24%) (Таблица 2). Единственными симптомами, с большей частотой обнаруживаемыми у пациентов с тЦФН по сравнению с пациентами без ЦФН, были пирамидные симптомы и нарушение слуха (18/24 (75%) vs 63/146 (43%) p = .004, и 6/24 (25%) vs 10/146 (7%) p = .013 соответственно). У трех пациентов с тЦФН и нарушениями слуха были выявлены митохондриальные заболевания, у остальных трех пациентов диагнозы отсутствовали. Даже среди пациентов с митохондриальными заболеваниями нарушения слуха чаще встречались при наличии тЦФН (3/4, 75%), нежели при отсутствии ЦФН (1/9, 11%; p = .005).
У одиннадцати пациентов с тЦФН определенный диагноз отсутствовал (таблица 2, столбец 4). У восьмерых из них симптоматика дебютировала в детском возрасте (71%), причем несколько неврологических симптомов совпало с симптомами, отмеченными у трех пациентов со взрослым дебютом симптоматики. В то же время лишь у пациентов с тЦФН отмечалась умственная отсталость (n = 6), эпилепсия (n = 3) и нарушения слуха (n = 3). Клинический фенотип пациентов с тЦФН и отсутствующим диагнозом отличался сложностью симптоматики. Так, среднее количество симптомов (исходя из списка из 13 симптомов, приведенного в таблице 2) среди таких пациентов составило 4.7 ( ± 1.6, 2–7) против 3.4 ( ± 1.7, 1–8) среди пациентов без ЦФН с отсутствием диагноза (p = .01).
3.3. Данные МРТ
У пациентов с тЦФН отмечались супратенториальные НЦБВ (нарушения целостности белого вещества; n = 8, 32%), общая атрофия (n = 7, 32%), НЦБВ в области задней черепной ямки (n = 4, 24%), нарушения базальных ганглиев (n = 4, 18%), и кальцификации мозговых структур (n = 3, 12%) (Таблица 2). У 23% (n = 5) пациентов с тЦФН не было отмечено отклонений на МРТ-снимках. Нарушения целостности белого вещества в области задней ямки и кальцификация областей мозга были единственными признаками, обнаруживавшимися чаще среди пациентов с тЦФН, нежели чем среди пациентов без ЦФН (4/17 (24%) vs 5/106 (5%), p = .021, и 3/25 (12%) vs 3/136 (2%), p = .048, соответственно), причем оба этих признака были ассоциированы в основном с наличием митохондриальных заболеваний, а кальцификации отмечались в основном среди пациентов с первичной кальцификацией головного мозга. Любопытно, что ни у одного из наших пациентов с митохондриальным заболеванием, не сопровождающимся ЦФН, не было отмечено ни кальцификаций, ни нарушений в белом веществе в области задней черепной ямки, в то время как среди пациентов с митохондриальными заболеваниями и тЦФН эти отклонения отмечались в 25% и 50% случаев соответственно.
Среди одиннадцати пациентов с тЦФН и без определенного диагноза основной находкой при МРТ-сканировании было наличие супратенториальных нарушений белого вещества – такие нарушения были отмечены у 8/11 человек (73%; 18% среди пациентов без ЦФН и без диагноза, p < .001).
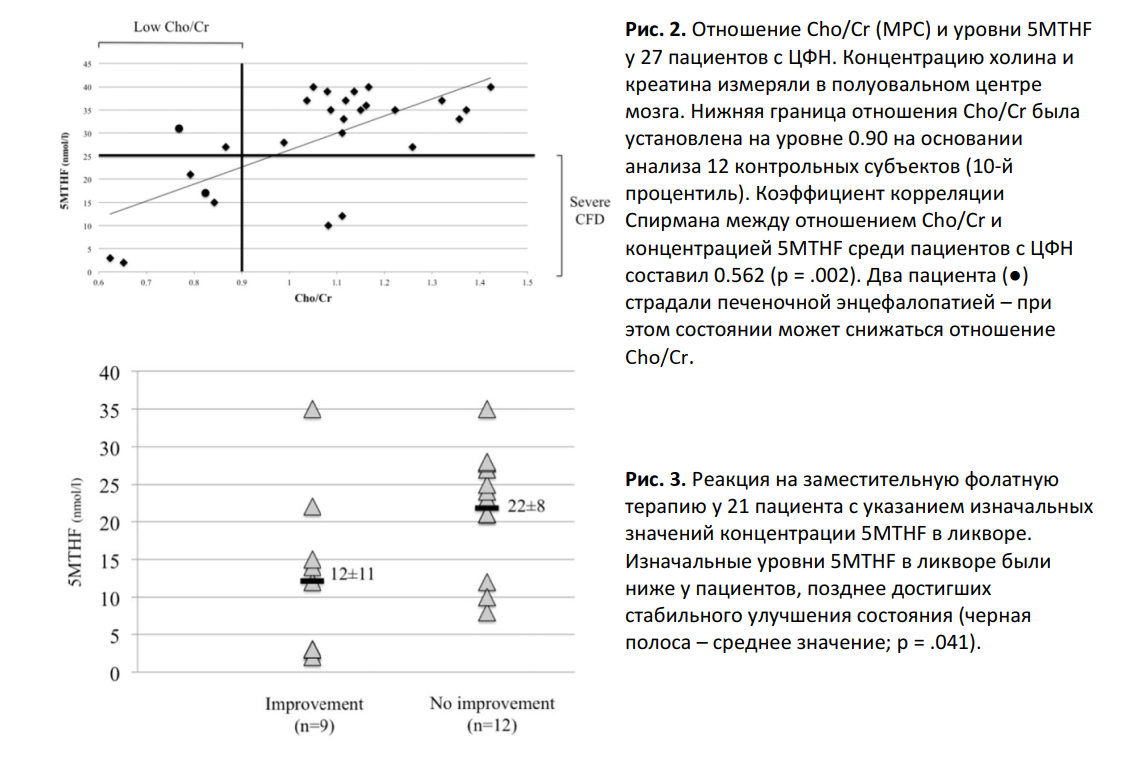
МРС-сканированию было подвергнуто 100 пациентов, в том числе 7 пациентов с тЦФН (из них у 5 пациентов не было определенного диагноза), 20 пациентов с нетяжелым ЦФН и 73 пациентов без ЦФН. Отношение Cho/Cr у пациентов было ниже, чем у членов контрольной группы (0.85 ± 0.19 vs 1.05 ± 0.13, p = .005) и чем у пациентов без ЦФН (0.85 ± 0.19, vs 1.11 ± 0.17, p = .005). Отношения NAA/Cr и Cho/NAA были одинаковыми во всех трех группах. В исследованной нами когорте чувствительность показателя «сниженное отношение Cho/Cr» в качестве параметра диагностики тЦФН составила 71%, а специфичность составила 92% (86/93). Специфичность достигала 97% в случае снижения диагностического предела (< 0.85 вместо < 0.90). Среди пациентов с ЦФР (n = 27) отмечалась значительная корреляция отношения Cho/Cr и концентрации 5MTHF (r = 0.562, p = .002) (Рис. 2).
3.4. Терапия и развитие болезни
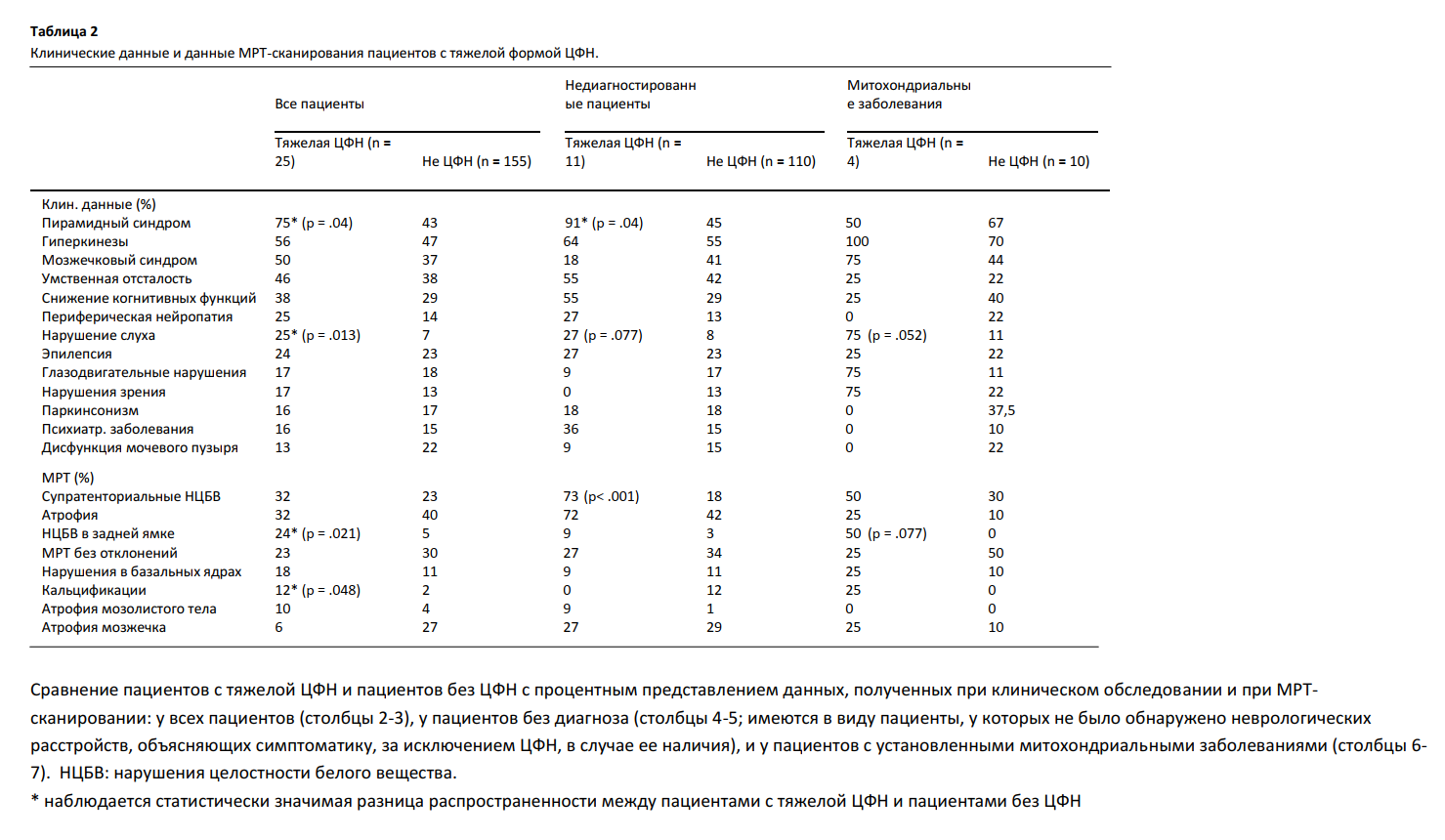
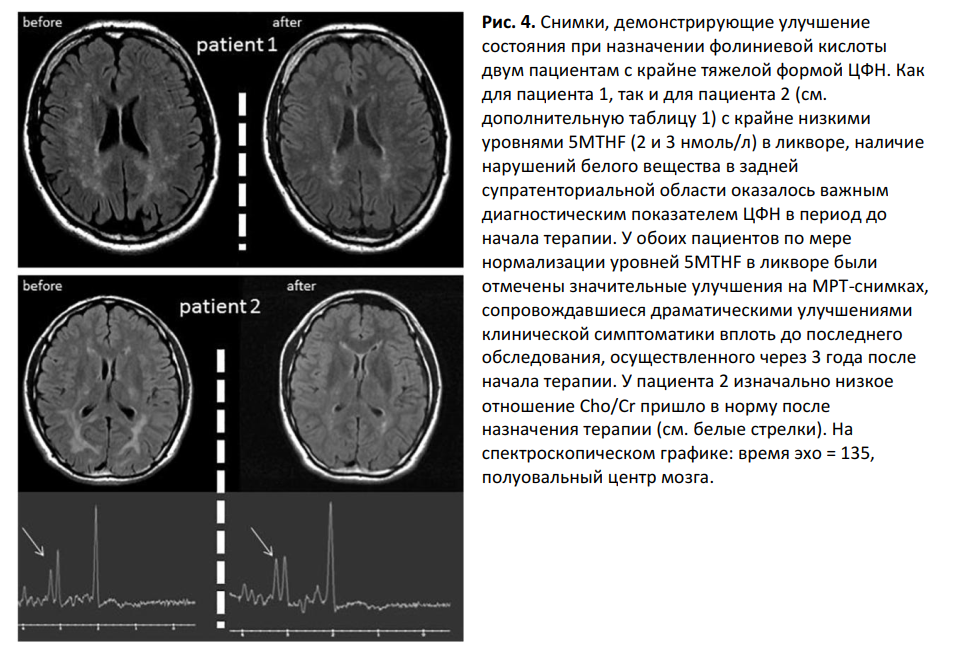
25 из 69 пациентов с ЦФН получали терапию фолатом в виде 5MTHF, фолиниевой кислоты, либо комбинации этих двух субстанций (дополнительная таблица 1). Фолиниевая кислота (25-75 мг/сутки) и 5MTHF продемонстрировали одинаковую эффективность. У всех пациентов, подвергшихся анализу ликвора после назначения заместительной фолатной терапии (n = 11), была отмечена нормализация уровней 5MTHF в ликворе. Среди 21 пациента, в отношении которых имелись данные последующего наблюдения, улучшение состояния было отмечено у 13 пациентов, при этом, согласно записям в медицинских картах, устойчивое улучшение состояния было достигнуто девятью пациентами. У пациентов с ЦФН, достигших устойчивого улучшения состояния, средняя концентрация 5MTHF в ликворе была изначально ниже, чем у пациентов, не достигших улучшения либо испытавших лишь временное улучшение состояния (12 ± 11 против 22 ± 8, p = .041) (Рис. 3). 8 из 16 (50%) пациентов с тЦФН, получавших терапию, достигли устойчивого улучшения состояния. Дебют симптоматики был отмечен в детском возрасте у 12 (63%) из 19 пациентов, получавших терапию, и у 7 (88%) из 8 пациентов, достигших стабильного улучшения состояния. Клинически заметное улучшение симптоматики выражалось в основном в улучшении походки (n = 7), снижении симптомов двигательных расстройств (n = 6) и улучшении поведения (n = 5). Два пациента без установленного диагноза, у которых отмечались крайне низкие концентрации 5MTHF в ликворе (2 и 3 нмоль/л) в сочетании с симметричными диффузными нарушениями структуры белого вещества и низким отношением Cho/Cr, продемонстрировали драматическое и устойчивое улучшение как клинических признаков, так и снимков мозга (рис. 4). Помимо описанного выше, у обоих пациентов не удалось выявить никаких отклонений в ходе обширного диагностического обследования, включавшего в себя полноэкзомное секвенирование. В частности, не было выявлено признаков митохондриальных заболеваний.
4. Обсуждение
В настоящей работе мы приводим данные клинических и радиологических исследований пациентов с диагнозом церебральной фолатной недостаточности, поставленным во взрослом возрасте, уделяя особое внимание 25 пациентам с тяжелой формой ЦФН (тЦФН). Это первое исследование столь крупной когорты взрослых пациентов, обследованных на предмет наличия ЦФН. Благодаря разнообразному составу пациентов нам удалось хорошо отразить картину проявлений ЦФН при неврологических заболеваниях. Однако в нашей работе, возможно, были недооценены некоторые ассоциированные с ЦФН состояния, при которых врачами не производилась оценка концентрации 5MTHF в ликворе ввиду достаточности имевшихся диагностических признаков. То, что среди пациентов наблюдался большой разброс возраста, в котором произошел дебют симптоматики, подчеркивает тот факт, что тЦФН не является состоянием, начинающимся исключительно в детском возрасте. ЦФН, диагностируемая во взрослом возрасте, вероятно, представляет собой состояние, отличное от ЦФН, описанной у детей. У исследованных нами пациентов не было выявлено наследуемых дефектов фолатного метаболизма (НДФМ). Более того, у недиагностированных пациентов с тЦФН в нашей выборке не наблюдалось фенотипа, характерного для идиопатической детской формы ЦФН – стоит особенно отметить, что у них часто отсутствовала эпилепсия [2,19]. Гетерогенность клинического фенотипа и МРТ-фенотипа у взрослых пациентов с тЦФН в нашей выборке, вероятно, свидетельствует о гетерогенности заболеваний, вызвавших недостаточность — будь то выявленных (например, митохондриальных заболеваний) либо оставшихся невыявленными. Вместе с тем в нашем исследовании выявлены важные особенности, позволяющие предположить, каких пациентов стоит включать в скрининг на наличие тЦФН в условиях клинической практики. Нам кажется, что кандидатами на такой скрининг должны быть пациенты с неврологическими заболеваниями неясной этиологии, особенно при наличии множества симптомов и/или диффузных симметричных нарушений структуры белого вещества на МРТ-снимках мозга и/или сниженного отношения Cho/Cr по данным МРС-сканирования. Возможность проведения скрининга также можно обсудить при наличии у пациента состояния, которое может привести к тЦФН – например, митохондриального заболевания, печеночной энцефалопатии, первичной кальцификации мозга, мутации SPG11 либо PGK1. Из девяти наших пациентов, продемонстрировавших устойчивое улучшение при назначении фолиниевой кислоты, у восьми была тяжелая форма ЦФН. В связи с этим мы рекомендуем назначение фолиниевой кислоты в качестве пробной терапии при уровне ниже 25 нмоль/л.
Как правило, у пациентов с тЦФН отмечался сложный фенотип с сочетанием различных неврологических симптомов. Пациенты с дебютом симптоматики после 15 лет были почти столь же многочисленны, как и пациенты с детским дебютом симптомов. Выраженная гетерогенность наблюдаемого фенотипа тЦФН, по-видимому, свидетельствует о том, что основные симптомы пациентов были скорее симптомами их исходного заболевания, нежели симптомами собственно ЦФН. Это предположение согласуется с тем, что у некоторых пациентов, несмотря на полную нормализацию уровней 5MTHF в ликворе под воздействием терапии, наблюдалось лишь частичное улучшение клинического состояния. Только три проявления были отмечены у ≥50% пациентов с тЦФН (пирамидный синдром, гиперкинезы и мозжечковый синдром). Высокая распространенность пирамидного синдрома напоминает о симптомах, наблюдаемых при НДФМ, например, при недостаточности фермента MTHFR [9]. Вместе с тем при таких дефектах часто развиваются периферическая нейропатия и эпилепсия, которые наблюдались у наших пациентов с тЦФН не так часто (в 25% и 24% случаев соответственно) – вероятно, потому что при НДФМ отмечается более выраженная ЦФН (< 5 нмоль/л, [29]). В то время как при НДФМ не отмечается снижение слуха, у наших пациентов с тЦФН снижение слуха было отмечено в 25% случаев, при этом лишь трое из этих пациентов страдали митохондриальными заболеваниями. Эти факты говорят в пользу того предположения, что тЦФН может представлять собой негативный фактор, способствующий ухудшению различных симптомов исходного заболевания, а особенно сильная церебральная фолатная недостаточность сама по себе вызывает некоторые симптомы. Вероятно, по этой причине наиболее яркие примеры эффективности терапии наблюдались у пациентов с глубокой фолатной недостаточностью, независимо от изначального неврологического диагноза.
Отклонения, наблюдаемые на МРТ-снимках при тЦФН, были гетерогенны и крайне различны по выраженности, вероятно, ввиду тех же причин, что вызывают гетерогенность клинических симптомов заболевания. Пациенты с тЦФН отличались от пациентов без ЦФН такими признаками, как нарушения структуры белого вещества в области задней ямки и наличие областей кальцификации. Диффузные симметричные нарушения белого вещества часто обнаруживаются у пациентов с поражением центральной нервной системы, особенно при метаболических заболеваниях. Невозможно установить степень вклада ЦФН в их развитие. И все же тот факт, что у двух пациентов монотерапия фолиниевой кислотой привела к улучшениям на снимках мозга, указывает на то, что при крайне низких уровнях 5MTHF в ликворе происходит повреждение белого вещества мозга – по крайней мере, в некоторых случаях. Обнаружение у пациентов с тЦФН кальцификаций мозга могло отражать наличие ПКМ-ассоциированных заболеваний либо митохондриальных заболеваний. Вместе с тем внутричерепные кальцификации описаны у пациентов с НДФМ (недостаточность FOLR1 [11] и наследственная мальабсорбция фолатов [30]), а также у пациентов с синдромом Айкарди-Гутьерес [31], что указывает на возможное наличие прямой связи с ЦФН. Метод МРС доказал свою пользу при диагностике нейрометаболических заболеваний и последующем наблюдении за пациентами, и он доступен в повседневной практике [32,33]. В ходе нашего исследования, пусть и проведенного на небольшой выборке, отношение Cho/Cr было значительно ниже у пациентов с тЦФН и коррелировало с выраженностью недостаточности. Печеночная энцефалопатия может вызывать ЦФН, а также ассоциирована с гипераммониемией – состоянием, при котором МРС-сканирование выявляет сниженную концентрацию холина [34]. Невозможно объяснить результаты МРС-сканирования в исследованной нами когорте наличием гипераммониемии, так как лишь у 2 из 7 пациентов с тЦФН и низким отношением Cho/Cr была выявлена печеночная энцефалопатия, причем у одного из них уровень аммиака не выходил за пределы нормы. Сниженное отношение Cho/Cr у пациентов с тЦФН может быть объяснено тем, что при ЦФН изменения процесса миелинизации ведут к недостаточной концентрации холина в глиальных клетках [2,13]. Ввиду того, что отношение Cho/Cr снижается из-за недостатка холина, можно предположить сохранение креатина на нормальном уровне. Это подразумевает сохранение нормального биосинтеза креатина в печени – процесса, использующего фолат для превращения гуанидинацетата в креатин посредством метилирования. Образующийся креатин переносится в мозг. Следовательно, сохранение креатина на нормальном уровне говорит в пользу того, что у большинства включенных в исследование пациентов уровни фолата в сыворотке были в пределах нормы. Чувствительность отношения Cho/Cr (около 71%) оказалась недостаточной для использования в отсутствие других инструментов диагностики, однако его высокая специфичность (92%) указывала на полезность этого показателя в качестве неинвазивного маркера тЦФН.
В ходе этого исследования тЦФН была выявлена лишь при нескольких заболеваниях: при печеночной энцефалопатии, митохондриальных заболеваниях, мутациях PGK1, мутациях SPG11, и при первичной кальцификации структур мозга. Сверх того, у семи пациентов были обнаружены антитела к FRα. Три механизма могут отвечать за связь этих состояний со сниженными уровнями 5MTHF в ликворе: (a) сбой метаболизма 5MHTF в мозге, (b) дисфункция транспортных молекул, переносящих 5MTHF, и (c) нарушение АТФ-зависимого переноса фолата в ЦНС. Существует гипотеза о том, что антитела к FRα у детей с неврологическими расстройствами и у взрослых пациентов с шизофренией могут нарушать перенос фолата в центральную нервную систему [2,20–22]. В нашей когорте пациенты с антителами к FRα (n = 7 из 9 обследованных) представляли собой гетерогенную группу как в плане возраста дебюта симптоматики (3 с дебютом в детстве, 2 – во взрослом возрасте, 2 – с неуточненным возрастом дебюта), так и в плане клинического фенотипа и концентрации 5MTHF в ликворе (среднее: 22.8, разброс 8-41). У двух пациентов, которым был проведен повторный анализ, наличие антител подтверждено не было – возможно, из-за такого известного явления, как колебание титра антител [21]. Трое пациентов получали иммуноглобулины внутривенно и/или проходили плазмаферез и не продемонстрировали улучшений, при этом все трое продемонстрировали как минимум преходящий ответ на восполняющую терапию фолиниевой кислотой. По этим причинам мы не можем сделать вывода относительно вклада аутоиммунных процессов в развитие ЦФН в нашей когорте. Согласно отчету о другом исследовании с участием более 67 детей с РАС (расстройствами аутистического спектра), включая 5 детей с ЦФН [35], антитела к FRα были обнаружены у 4 пациентов без ЦФН, что ставит под сомнение патофизиологическую роль таких антител в некоторых популяциях. Что же касается митохондриальных заболеваний, они вызывают энергетический сбой, предположительно нарушающий АТФ-зависимый перенос фолата в ЦНС. Вдобавок к этому при синдроме Кернса-Сейра анатомические и физиологические изменения в сосудистом сплетении также могут нарушать перенос 5MTHF в ЦНС, и это может объяснять повышенную концентрацию белков в ликворе, наблюдаемую в типичных случаях заболевания [17]. У наших пациентов с тЦФН, в том числе пациентов без установленного причинного заболевания, средняя концентрация общего белка в ликворе (N < 0.35) была повышена (0.79 г/л ± 0.58 [0.12–2.12]), позволяя предположить, что, как и в случае с синдромом Кернса-Сейра, развитие церебральной фолатной недостаточности можно объяснить сбоем механизма переноса фолата через сосудистое сплетение.

Также при митохондриальных заболеваниях в мозге происходит индукция процессов окислительного стресса, что может ускорять катаболизм 5MTHF [36]. Согласно полученным нами результатам, тяжелая ЦФН может свидетельствовать о наличии у пациента митохондриального заболевания, и врачу в таких случаях следует расследовать эту гипотезу. В случаях печеночной энцефалопатии наличие ЦФН нельзя объяснить лишь ролью печени в метаболизме фолатов, поскольку ни у одного из наших пациентов с ПЭ не было выявлено снижения концентрации фолата в плазме крови. При ПЭ отмечается измененное протекание цикла Кребса, которое может, вызывая нарушение энергетического обмена, приводить к ЦФН [37]. ФГК1 (недостаточность фосфоглицераткиназы тип 1) – редкое X-сцепленное заболевание, классически проявляющееся гемолитической анемией и миопатией; у исследованного нами пациента, однако, была отмечена редкая картина проявлений, хотя и описанная в литературе, напоминающая митохондриальное заболевание и включающая в себя умственную отсталость, паркинсонизм, пигментный ретинит и инсультно-подобные эпизоды [38]. Ассоциацию заболевания с тЦФН можно объяснить энергетической недостаточностью, особенно с учетом того, что при недостаточности ФГК отмечается не только изменение процесса гликолиза, но и митохондриальные изменения [39]. Два пациента с мутациями гена SPG11 (спастическая параплегия 11 - заболевание, при котором отмечается умственная отсталость, спастический парапарез, периферическая моторная нейропатия и истончение мозолистого тела [40]) не получали терапии фолатом. Любопытно, что затронутый мутацией при СПГ11 белок спатаксин частично экспрессирован в митохондриях, в которых он может играть роль рецептора или транспортера, что согласуется с данными, полученными в отношении других форм наследственной спастической параплегии, и подчеркивает возможную роль митохондрий в заболевании [41,42]. Следовательно, и в данном случае энергетический дефект может являться патофизиологическим механизмом, приводящим к ЦФН.
Дополнительные данные к этой статье размещены онлайн: https:// doi.org/10.1016/j.jns.2018.11.014.
Соответствие этическим нормам и получение согласия на участие
Авторы заявляют, что получили все необходимые одобрения, связанные с соответствием этическим нормам, и что все пациенты, участвовавшие в исследовании, предоставили свое согласие.
Согласие на публикацию
Неприменимо (рукопись не содержит персональных данных).
Доступность данных и материалов
С просьбами о дополнительной информации обращайтесь к авторам.
Возможные конфликты интересов
Авторы заявляют об отсутствии столкновения интересов финансового либо иного плана в отношении данной рукописи.
Финансирование
Финансирования не потребовалось.
Вклад авторов
Авторы MM и YN работали над концепцией и планом исследования, занимались анализом и интерпретацией данных и составлением первой черновой версии рукописи. Авторы YN, CL, FS и ER занимались получением клинических данных, а JFB и FM – получением биохимических данных. Все авторы ознакомились с окончательным вариантом рукописи и одобрили его.
Благодарности
Мы выражаем благодарность следующим врачам за направление пациентов на исследование: Isabelle An, Perrine Charles, Sophie Dupont, Claire Ewenczyk, Anne Francheschini Mandel, Isabelle Le Ber, Fanny Mochel, Mathieu Anheim, Bertrand Degos, David Grabli, Pascal Lebray, Vincent Meininger, Joseph Moussalli.
Список литературы
[1] V. Ramaekers, N. Blau, Cerebral folate deficiency, Dev. Med. Child Neurol. 46 (2004) 843–851.
[2] V. Ramaekers, J.M. Sequeira, E.V. Quadros, Clinical recognition and aspects of the cerebral folate deficiency syndromes, Clin. Chem. Lab. Med. 51 (2013) 497–511.
[3] K. Hyland, J. Shoffner, S.J. Heales, Cerebral folate deficiency, J. Inherit. Metab. Dis. 33 (5) (2010) 563–570.
[4] H.J. Blom, Y. Smulders, Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects, J. Inherit. Metab. Dis. 34 (2011) 75–81.
[5] M. Serrano, B. Pérez-Dueñas, J. Montoya, A. Ormazabal, R. Artuch, Genetic causes of cerebral folate deficiency: clinical, biochemical and therapeutic aspects, Drug Discov. Today 17 (2012) 1299–1306.
[6] R. Spector, Nutrient transport systems in brain: 40 years of progress, J. Neurochem. 111 (2009) 315–320.
[7] R. Spector, C.E. Johanson, Vectorial ligand transport through mammalian choroid plexus, Pharm. Res. 27 (2010) 2054–2062.
[8] A. Torres, S.A. Newton, B. Crompton, et al., CSF 5-Methyltetrahydrofolate serial monitoring to guide treatment of congenital folate malabsorption due to proton-coupled folate transporter (PCFT) deficiency, JIMD Rep. 24 (2015) 91–96.
[9] H. Cario, D.E. Smith, H. Blom, et al., Dihydrofolatereductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folatedefi-cicency leading to severe neurologic disease, Am. J. Hum. Genet. 88 (2011)226–231.
[10] A. Gales, M. Masingue, S. Millecamps, S. Giraudier, L. Grosliere, C. Adam, et al., Adolescence/adult onset MTHFR deficiency may manifest as isolated and treatable distinct neuro-psychiatric syndromes, Orphanet J. Rare Dis. 13 (1) (2018) 29.
[11] S.P. Toelle, D. Wille, B. Schmitt, I. Scheer, B. Thöny, B. Plecko, Sensory stimulus-sensitive drop attacks and basal ganglia calcification: new findings in a patient with FOLR1 deficiency, Epilept. Disord. 6 (2014) 88–92.
[12] B. Perez-Dueñas, C. Toma, A. Ormazábal, et al., Progressive ataxia and myoclonic epilepsy in a patient with a homozygous mutation in the FOLR1 gene, J. Inherit. Metab. Dis. 33 (2010) 795–802.
[13] R. Steinfeld, M. Grapp, R. Kraetzner, et al., Folate receptor Alpha defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder asso-ciated with disturbed myelin metabolism, Am. J. Hum. Genet. 85 (2009) 354–363.
[14] F. Delmelle, B. Thöny, P. Clapuyt, N. Blau, M.C. Nassogne, Neurological improve-ment following intravenous high-dose folinic acid for cerebral folate transporter deficiency caused by FOLR-1 mutation, Eur. J. Paediatr. Neurol. 20 (5) (2016 Sep) 709–713.
[15] M. Pineda, A. Ormazabal, E. López-Gallardo, et al., Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion, Anal. Neurol. 59 (2006) 394–398.
[16] M. Serrano, M.T. García-Silva, E. Martin-Hernandez, et al., Kearns-Sayre syndrome: cerebral folate deficiency, MRI findings and new cerebrospinal fluid biochemical features, Mitochondrion 10 (2010) 429–432.
[17] K. Tanji, E.A. Schon, S. Dimauro, E. Bonilla, Kearns-sayre syndrome: oncocytic transformation of choroid plexus epithelium, J. Neurol. Sci. 178 (2000) 29–36.
[18] A. Ho, D. Michelson, G. Aaen, S. Ashwal, Cerebral folate deficiency presenting as adolescent catatonic schizophrenia : a case report, J. Child Neurol. 25 (2010)898–900.
[19] S. Mangold, N. Blau, T. Opladen, et al., Cerebral folate deficiency: a neurometabolic syndrome? Mol. Genet. Metab. 104 (2011) 369–372.
[20] V.T. Ramaekers, S.P. Rothenberg, J.M. Sequeira, et al., Autoantibodies to folate receptors in the cerebral folate deficiency syndrome, N. Engl. J. Med. 352 (2005) 1985–1991.
[21] V.T. Ramaekers, B. Thöny, J.M. Sequeira, et al., Folinic acid treatment for schizo-phrenia associated with folate receptor autoantibodies, Mol. Genet. Metab. 113 (2014) 307–314.
[22] J.M. Sequeira, V.T. Ramaekers, E.V. Quadros, The diagnostic utility of folate re-ceptor autoantibodies in blood, Clin. Chem. Lab. Med. 51 (2013) 545–554.
[23] F.J. Hansen, N. Blau, Cerebral folate deficiency: life-changing supplementation with folinic acid, Mol. Genet. Metab. 84 (2005) 371–373.
[24] P. Ferreira, S.M. Luco, S.L. Sawyer, J. Davila, K.M. Boycott, D.A. Dyment, Late diagnosis of cerebral folate deficiency: fewer seizures with folinic acid in adult siblings, Neurol. Genet. 2 (2015) e38.
[25] Z. Sadighi, I.J. Butler, M.K. Koenig, Adult-onset cerebral folate deficiency, Arch. Neurol. 69 (2012) 778–779.
[26] A. Ormazabal, A. García-Cazorle, B. Pérez-Dueñas, et al., Determination of 5-me-thyltetrahydrofolate in cerebrospinal fluid of paediatric patients: reference values for a paediatric population, Clin. Chim. Acta 371 (2006) 159–162.
[27] Verbeek, M., Blom, M., Wevers, R., Lagerwerf, J., van de Geer, J., Willemsen, M.P. Technical and biochemical factors affecting cerebrospinal fluid 5-MTHF, biopterin and neopterin concentrations. Mol. Genet. Metab., 2008;95:127–32.
[28] N. Blau, T. Opladen, Laboratory guide to the methods in biochemical genetics, in: N. Blau, M. Duran, K.M. Gibsons (Eds.), Folates, Springer-Verlag Berlin Heidelberg, 2008, pp. 717–724.
[29] B. Perez-Dueñas, A. Ormazábal, C. Toma, et al., Cerebral folate deficiency syn-dromes in childhood: clinical, analytical, and etiologic aspects, Arch. Neurol. 68 (2011) 615–621.
[30] I. Ahmad, G. Mukhtar, J. Iqbal, S.W. Ali, Hereditary folate malabsorption with extensive intracranial calcification, Indian Pediatr. 52 (2015) 67–68.
[31] R. La Piana, C. Uggetti, F. Roncarolo, et al., Neuroradiologic patterns and novel imaging findings in Aicardi-Goutières syndrome, Neurology 86 (2016) 28–35.
[32] D.J. Warren, D.J. Connolly, I.D. Wilkinson, M.J. Sharrard, P.D. Griffiths, Magnetic resonance spectroscopy changes following haemopoietic stem cell transplantation in children with cerebral adrenoleukodystrophy, Dev. Med. Child Neurol. 49 (2007) 135–139.
[33] S. Seidel, G. Kasprian, D. Prayer, M. Krssák, T. Sycha, E. Auff, Visualisation of treatment response in a case of cerebrotendinousxanthomatosis, J. Neurol. Neurosurg. Psychiatry 82 (2011) 703–704.
[34] A. Rovira, J. Alonso, J. Córdoba, MR imaging findings in hepatic encephalopathy, Am. J. Neuroradiol. 29 (2008) 1612–1621.
[35] J. Shoffner, B. Trommer, A. Thurm, et al., CSF concentrations of 5-methyltetrahy-drofolate in a cohort of young children with autism, Neurology 86 (2016) 2258–2263.
[36] A. Ormazabal, M. Casado, M. Molero-Luis, et al., Can folic acid have a role in mi-tochondrial disorder? Drug Discov. Today 20 (2015) 1349–1354.
[37] N. Weiss, P. Barbier Saint Hilaire, B. Colsch, et al., Cerebrospinal fluid metabo-lomics highlights dysregulation of energy metabolism in overt hepatic encephalo-pathy, J. Hepatol. 65 (2016) 1120–1130.
[38] Beutler E.P.G.K. Deficiency, Br. J. Haematol. 136 (2006) 3–11.
[39] J.M. Schröder, R. Dodel, J. Weis, et al., Mitochondrial changesin muscle phos-phoglycerate kinase deficiency, Clin. Neuropathol. 15 (1996) 34–40.
[40] G. Stevanin, H. Azzedine, P. Denora, et al., Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline, and lower motor neuron degeneration, Brain 131 (2008) 772–784.
[41] G. Stevanin, F.M. Sabtorelli, H. Azzedine, et al., Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum, Nat. Genet. 39 (2007) 366–372.
[42] A.H. Crosby, C. Proukakis, Is the transportation highway the right road for her-editary spastic paraplegia? Am. J. Hum. Genet. 71 (2002) 1009–1016.