Возможная причина фармакорезистентной эпилепсии у ребенка: церебральная фолатная недостаточность, болезнь, поддающаяся терапии
Авторы: Sarah Mafi 1, Cécile Laroche-Raynaud 2,3, Pauline Chazelas 1,4, Anne-Sophie Lia 1,4,5, Paco Derouault 5, Franck Sturtz 1,4, Yasser Baaj 1, Rachel Froget 2,6, Marlène Rio 7, Jean-François Benoist 8, François Poumeaud 4, Frédéric Favreau 1,4 и Pierre-Antoine Faye 1,4,*
(скачать этот перевод в формате PDF)
1 Университетская больница города Лимож, Отделение биохимии и молекулярной генетики (CHU de Limoges, Service de Biochimie et Génétique Moléculaire, F-87000 Limoges, France)
2 Университетская больница города Лимож, Отделение педиатрии (CHU de Limoges, Service de Pédiatrie, F-87000 Limoges, France)
3 Университетская больница города Лимож, Центр компетенций наследственных заболеваний метаболизма (CHU de Limoges, Centre de Compétence des Maladies Héréditaires du Métabolisme, F-87000 Limoges, France)
4 Факультет медицины университета города Лимож, Лаборатория EA6309 "Поддержание миелиновой оболочки и миелин-опосредованные нейропатии" (Faculté de Médecine, EA6309 Maintenance Myélinique et Neuropathies Périphériques, Université de Limoges, F-87000 Limoges, France)
5 Университетская больница города Лимож, Факультет биоинформатики (CHU Limoges, UF de Bioinformatique, F-87000 Limoges, France)
6 Университетская больница города Лимож, Центр клинических исследований при Национальном институте здоровья и медицинских исследований INSERM (CHU Limoges, INSERM CIC 1435, F-87000 Limoges, France)
7 Университетская детская больница Некер, Париж, Отделение генетики (CHU Necker, Enfants Malades, Paris, APHP, Service de Génétique, F-75743 Paris, France)
8 Университетская детская больница Некер, Париж, Отделение метаболомной биохимии (CHU Necker, Enfants Malades, Paris, APHP, Service de Biochimie Métabolomique, F-75743 Paris, France)
* Автор, ответственный за переписку.
Brain Sci. 2020, 10(11), 762; https://doi.org/10.3390/brainsci10110762
Рукопись получена: 25 сентября 2020 г./ Пересмотрена: 16 октября 2020 г./ Принята: 19 октября 2020 г./ Опубликована: 22 октября 2020 г.
Аннотация
Церебральная фолатная недостаточность (ЦФН) – неврологическое заболевание, для которого характерно снижение концентрации 5-метилтетрагидрофолата (5-MTHF) в спинномозговой жидкости (СМЖ). Оценочная распространенность этого заболевания, наследуемого по аутосомно-рецессивному типу, составляет менее 1 человека на миллион. На данный момент описано пятнадцать патогенных вариаций гена FOLR1 (фолатный рецептор 1), кодирующего фолатный рецептор альфа (FRα). Мы описываем новую патогенную вариацию гена FOLR1 у пациентки детского возраста. Нашей целью было описание основной структуры белка FRα, необходимой для его работы. В больницу поступил ребенок трехлетнего возраста с первичным эпизодом фебрильных судорог. Через несколько месяцев были отмечены не сопровождавшиеся лихорадкой эпилептические приступы, а также снижение двигательных навыков и когнитивных способностей. Назначение различных противоэпилептических препаратов не позволяло контролировать приступы. На МРТ-снимках наблюдалась центральная гипомиелинизация, а биологический анализ выявил значительное снижение концентрации 5-MTHF в СМЖ. Секвенирование методами нового поколения (NGS) подтвердило наличие гомозиготной вариации (c.197 G > A, p.Cys66Tyr) гена FOLR1, вызывающей ЦФН. Данная вариация приводит к изменению белковой структуры фолатного рецептора альфа и указывает на важность дисульфидной связи Cys66-Cys109, необходимой для переноса 5-MTHF в ЦНС. К счастью, наш пациент с тяжелой формой ЦФН исключительно хорошо прореагировал на назначение больших доз фолиниевой кислоты перорально и внутривенно.
Ключевые слова: церебральная фолатная недостаточность; вариация гена FOLR1; нейродегенеративное заболевание; эпилепсия; кристаллографически определенная структура белка; педиатрия
-
Введение
Фолаты, или витамин B9 (B9) являются необходимым элементом для множества биологических процессов, таких как синтез и репарация ДНК, регулирование экспрессии генов, метаболизм аминокислот (АК), образование миелина и синтез нейромедиаторов [1]. Также хорошо известна их роль в формировании нервной трубки в ходе эмбриогенеза [2].
Несмотря на присутствие фолатов во множестве продуктов питания [3], у человека может развиться недостаточность фолата в организме. Усвоение фолатов происходит в кишечнике, после чего в печени происходит их преобразование в 5-метилтетрагидрофолат (5-MTHF). Фолаты – семейство витаминов группы B, потребляемых человеком с пищей в виде фолиниевой кислоты (5-формил-ТГФ), конвертируемой в 5-MTHF, а также принимаемых в синтетической форме (фолиевая кислота). В организме фолиевая кислота восстанавливается до 7,8-дигидрофолата (DHF), затем до 5,6,7,8-тетрагидрофолата в реакции, катализируемой ферментом дигидрофолатредуктазой (DHFR) [4]. Одноуглеродный фрагмент переносится от серина к THF ферментом серингидроксиметилтрансферазой (SHMT). В ходе этой реакции образуются глицин и 5,10-метилентетрагидрофлат, участвующий в синтезе и репарации нуклеиновых кислот. Последний затем преобразуется в 5-MTHF с помощью фермента метилентетрагидрофолатредуктазы (MTHFR). Фолиниевая кислота либо непосредственно участвует в метаболических процессах, либо подвергается преобразованию в 5,10-метилен-ТГФ в двух последовательных реакциях с участием ферментов 5,10-метенилтетрагидрофолатсинтетаза и метенилтетрагидрофолатдегидрогеназа [5]. В процессе преобразования 5-MTHF в THF высвобождается метильная группа. Эта группа используется для реметилирования гомоцистеина – реакции, продуктом которой является метионин, служащий прекурсором S-аденозилметионина (SAM). SAM – универсальный донор одноуглеродных групп, участвующий в процессах метилирования ДНК, РНК, липидов, белков и нейромедиаторов [6] (рисунок 1).
![]()
Рисунок 1. Метаболизм фолиевой кислоты и перенос 5-MTHF через сосудистое сплетение в ткани мозга. Красными стрелками и крестиками обозначены последствия нарушения работы фолатного рецептора альфа. Синими стрелками обозначены последствия назначения фолиниевой кислоты. Сокращения: 5-MTHF – 5-метилентетрагидрофолат; B6 – витамин B6; B12 – витамин B12; DHFR – дигидрофолатредуктаза; FRα – фолатный рецептор альфа; Gly – глицин; GSH – глутатион в восстановленном состоянии; GSSG – глутатион в окисленном состоянии; MS – метионинсинтаза; MTHFD – метилентетрагидрофолатдегидрогеназа; MTHFR – метилентетрагидрофолатредуктаза; MTHFS – метилентетрагидрофолатсинтетаза; PC – фосфатидилхолин; PCFT – протон-сопряженный транспортер фолатов; PI – фосфатидилинозитол; RFC – переносчик восстановленных фолатов; SAH – S-аденозилгомоцистеин; SAM – S-аденозилметионин; Ser – серин; SHMT – серингидроксиметилтрансфераза; SM – сфингомиелин.
В центральной нервной системе присутствуют различные системы транспорта фолата, в том числе переносчик восстановленных фолатов (RFC), протон-сопряженный транспортер фолатов (PCFT) и фолатный рецептор альфа (FRα). FRα обладает повышенной аффинностью к фолату по сравнению с RFC и PCFT. Перенос 5-MTHF в мозг в основном осуществляется FRα и PCFT сквозь базолатеральную мембрану эпителиальных клеток сосудистого сплетения, а затем FRα и RFC через апикальную щеточную каемку клеток [6]. Работа данных транспортеров, участвующих в перемещении 5-MTHF через гематоэнцефалический барьер, еще не изучена полностью, однако есть описания некоторых механизмов, предположительно задействованных в переносе фолата в мозг. Согласно Grapp et al. и Zhao et al., при достижении сосудистого сплетения 5-MTHF связывается с рецепторами FRα на базолатеральной стороне клеток. Комплексы 5MTHF-FRα попадают внутрь клеток в процессе рецепторно-опосредованного эндоцитоза. После попадания в клетку образовавшиеся везикулы перемещаются в ГФИ-заякоренные обогащенные белком ранние эндосомальные компартменты (GEEC). GEEC-компартменты переносятся к созревшим эндосомам - мультивезикулярным комплексам (MVC). Интралюминальные везикулы (ILV), содержащие FRα-связанные комплексы, накапливаются внутри MVC-везикул, а затем высвобождаются, уже в виде экзосом, из клетки в спинномозговую жидкость при слиянии MVC-везикул с апикальной стороной эпителия. За этим следует их эндоцитоз клетками эпендимы [7, 8]. По данным Requena Jimenez et al., в этих условиях важную роль играет фермент 10-формил-ТГФ-дегидрогеназа (FDH), обнаруживаемый совместно с 5-MTHF и FRα в эндоцитарных везикулах. В in-vitro исследованиях было продемонстрировано, что фермент FDH связывается с рецептором FRα и участвует в регулировке переноса фолатов, контролируя флуктуации уровней 5-MTHF. Концентрация фермента FDН в спинномозговой жидкости также может играть ключевую роль в созревании паутинной оболочки мозга [9].
Церебральная фолатная недостаточность (ЦФН) – редкий неврологический синдром (OMIM #613068), при котором в спинномозговой жидкости (СМЖ) отмечается недостаток 5-MTHF. Описан ряд причин, нарушающих работу рецептора FRα, например, митохондриальные заболевания, мутации гена, кодирующего фолатный рецептор альфа (FOLR1) [10], и, возможно, наличие аутоантител к рецептору FRα [11]. Ген FOLR1 расположен на хромосомном участке 11q13.4 и содержит семь экзонов. Описано как минимум пятнадцать патогенных вариантов гена, приводящих к производству дефектного белка [6].
-
Описание клинического случая
Работа была одобрена Комитетом по этике Университетской больницы Лиможа (разрешение № 364-2020-20). Более того, было получено согласие обоих родителей на распространение полученных данных. Настоящее исследование было проведено в согласии с Хельсинской декларацией. В детское реанимационное отделение поступила девочка трехлетнего возраста с первичным эпизодом фебрильных судорог. Пациент – единственный ребенок родителей, не связанных кровным родством. Девочка родилась в срок с нормальным весом и нормальной жизнеспособностью. Первые 18 месяцев показатели психомоторного развития оставались в рамках нормы, после чего у девочки развилась атактическая походка, тремор, раздражительность, расстройства сна и нарушения речи. Лечение эпилептического эпизода заключалось в ректальном введении диазепама. Причиной судорог была сочтена лихорадка (38.5 °C).
Через 10 месяцев случился второй эпизод эпилептических приступов, за которым в последующие месяцы были отмечены новые эпизоды, не сопровождавшиеся повышением температуры. Было осуществлено МРТ-сканирование мозга, выявившее тяжелую гипомиелинизацию, отражавшуюся в усилении сигналов в субкортикальных структурах белого вещества, особенно в области мозжечка и субтенториальном пространстве, преимущественно сзади (рисунок 2). Электронейромиография не выявила признаков поражения периферических нервов. Анализ хромосом методом кариотипирования и матричная сравнительная геномная гибридизация не выявили отклонений. Диагностика слуха и офтальмологическое обследование также не выявили проблем. Оценивались биологические показатели плазмы крови (аммиак, пируват, креатинкиназа, ацилкарнитиновый профиль, сиалотрансферриновый профиль, аминокислоты, фитановая кислота, пристановая кислота, жирные кислоты с очень длинной цепью), мочи (аминокислоты, профиль органических кислот, сульфатиды, мукополисахариды, лейкоцитарные ферменты) и спинномозговой жидкости (аминокислоты). Все показатели были в пределах нормы.
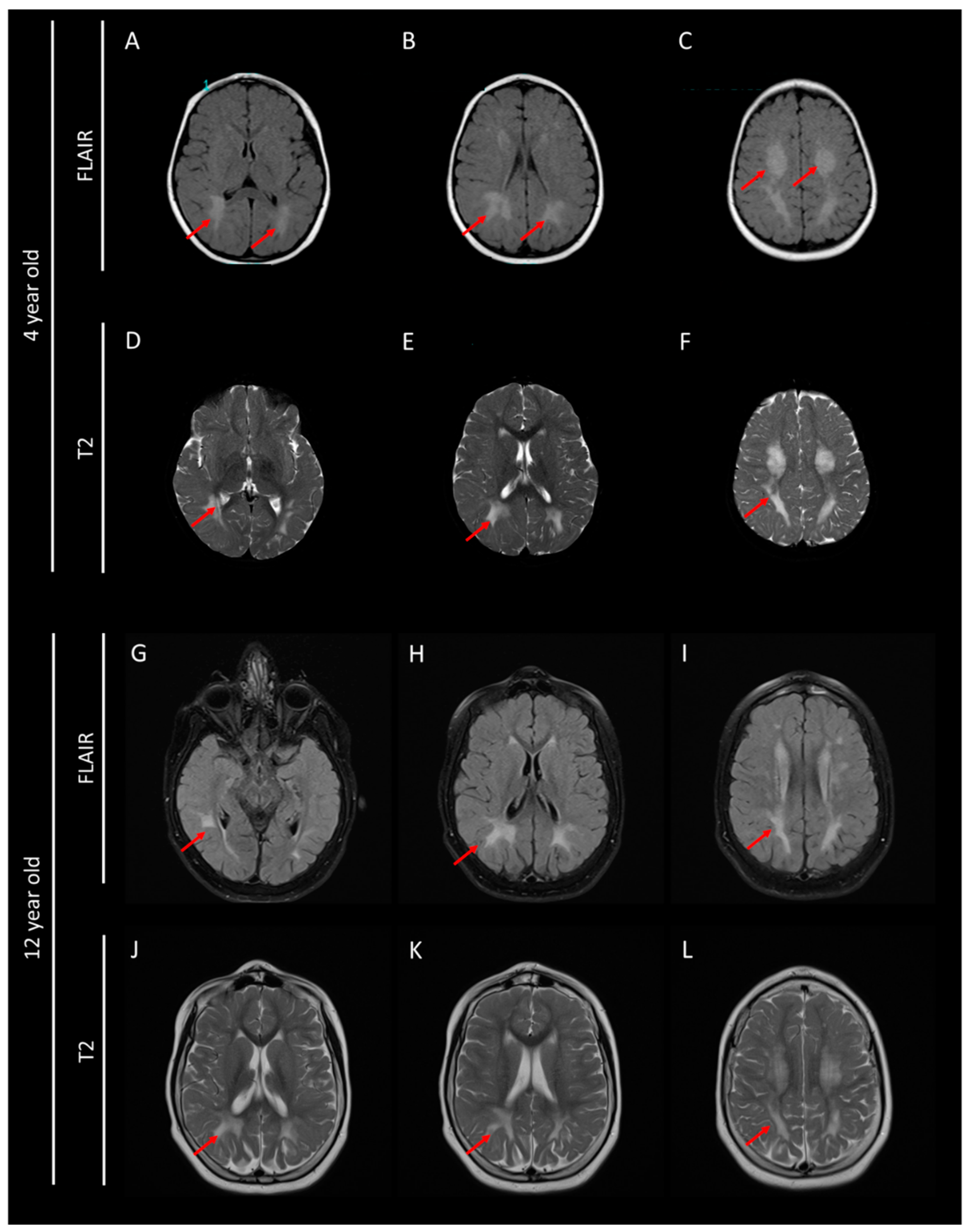
Рисунок 2. Результаты МРТ-сканирования мозга в возрасте 4 и 12 лет с использованием последовательностей FLAIR (A–C,G–I) и T2 (D–F,J–L). Красными стрелками указаны диффузные изменения белого вещества, связанные с гипомиелинизацией.
В возрасте 5 лет было отмечено увеличение частоты приступов. КТ-сканирование выявило билатеральные области кальцификации в бледном шаре и подкорковых структурах теменной и затылочной долей (рисунок 3). На электроэнцефалограмме была отмечена медленноволновая активность и комплекс пик-волна, ассоциированный с эпилептиформной активностью, в особенности в областях гипомиелинизации. Были опробованы различные комбинации антиэпилептических препаратов, которые изначально назначались по схеме монотерапии, а затем битерапии, включая клобазам, вальпроат натрия, ламотриджин, леветирацетам, топирамат, пирацетам и зонисамид. Антиэпилептическая терапия оказалась неэффективной и сопровождалась побочными эффектами, в особенности при назначении клобазама, топирамата и пирацетама.
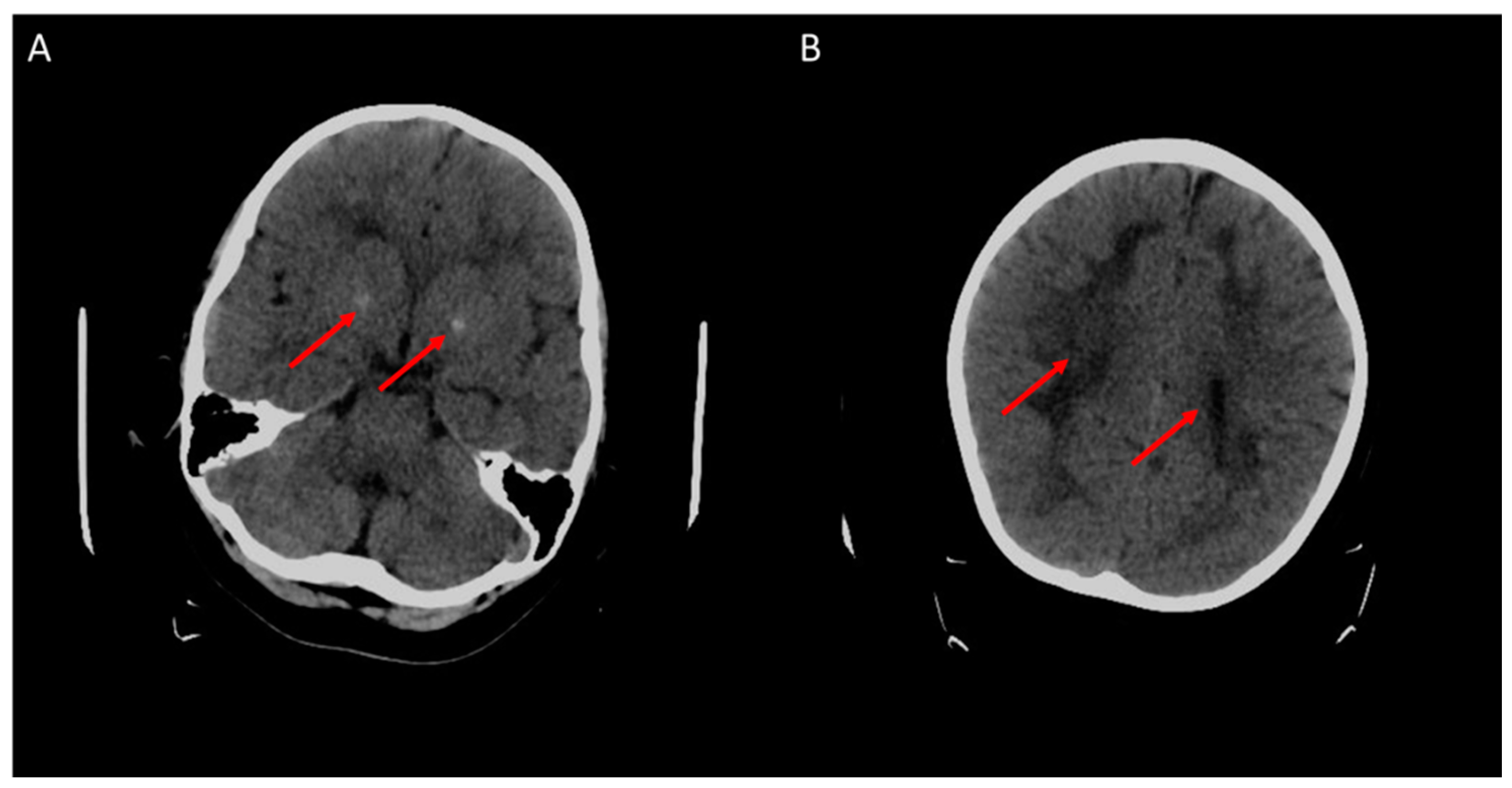
Рисунок 3. КТ-снимки мозга, сделанные в возрасте 4 лет. Красными стрелками указаны очаги кальцификации (A) и диффузных изменений белого вещества (B).
Происходило постепенное усугубление дискинезии, атрофии мозжечка и клинических проявлений энцефалопатии. В возрасте около 11 лет было отмечено постепенное снижение речевых и двигательных навыков и пациентка потеряла способность ходить без посторонней помощи. Не реже раза в день наблюдались миоклонические приступы, сопровождавшиеся приступами, вероятно, атонического характера.
Было предпринято тщательное диагностическое исследование показателей метаболизма. Подозрение на ЦФН возникло по результатам анализа спинномозговой жидкости, в которой была выявлена очень низкая концентрация 5-MTHF (1 нмоль/л; референсные значения: > 44 нмоль/л) вкупе с необычно низкой концентрацией фолата в плазме крови (<2 нг/мл; референсные значения: 3.89–26.8 нг/мл), несмотря на нормальные уровни фолата в эритроцитах. Первоначально врачи исключали наличие диетической недостаточности фолата ввиду нормальных концентраций гомоцистеина и витамина B12 (B12) в плазме пациентки, нормального количества эритроцитов и нормального содержания фолата в эритроцитах, а также соблюдения сбалансированной диеты, по данным родственников. Остальные показатели метаболизма не дали существенной информации (таблица 1). Были задействованы методики секвенирования нового поколения, чтобы проанализировать панель генов, связанных с развитием церебральной фолатной недостаточности (приложение A). Была получена библиотека ДНК с помощью процедуры Sureselect (Agilent) для исследования экзонов и экзон-интронных границ (+/−50 п.о.). Секвенирование проводили с помощью секвенатора NextSeq500 (Illumina). Было достигнуто 100%-е 30-кратное покрытие, а средства биоинформатики позволили осуществить выявление SNV (однонуклеотидных вариантов) и CNV (вариаций числа копий). Диагноз церебральной фолатной недостаточности был подтвержден благодаря выявлению новой гомозиготной замены c.197 G > A (p.Cys66Tyr) в экзоне 4. Использование панели генов, приспособленной для работы с NGS-методами, не выявило иных патогенных вариаций. Как и ожидалось, оба родителя оказались гетерозиготными носителями выявленного варианта.
Таблица 1. Метаболический профиль спинномозговой жидкости (СМЖ) до назначения терапии.
|
Показатели |
Концентрация в образцах СМЖ пациента |
Референсный диапазон |
|
Лактат |
0.40 |
<2.20 ммоль/л |
|
Пируват |
0.05 |
<0.14 ммоль/л |
|
Отношение лактат/пируват |
7.9 |
<20 |
|
Ацетоуксусная кислота |
0.02 |
|
|
Бета-гидроксимасляная кислота |
<0.01 |
|
|
3-O-метилдофа |
10.5 |
3–54 нмоль/л |
|
3-метокси-4-гидроксифенилгликоль |
19.6 |
11–46 нмоль/л |
|
5-гидрокситриптофан |
8.6 |
3–12 нмоль/л |
|
5-гидроксииндолуксусная кислота (HIAA) |
88 |
63–185 нмоль/л |
|
Гомованилиновая кислота |
231 |
156–410 нмоль/л |
|
Биоптерин |
10.8 ↓ |
14–36 нмоль/л |
|
Неоптерин |
10.2 |
10–24 нмоль/л |
|
5-метилтетрагидрофолат |
1↓↓↓ |
>44 нмоль/л |
|
Интерферон альфа |
<2 |
<2 МЕ/мл |
Сокращения: ↓: снижение; ↓↓↓: сильное снижение.
Вариация, описанная нами, отсутствовала в архиве ClinVar и была обнаружена в единственном экземпляре среди 251476 аллелей, содержащихся в базе данных GnomAD (версия 2.1.1) [12]. Любопытно, что программа, широко используемая для оценки патогенности, оценила данный вариант как обладающий высокой степенью патогенности (таблица 2). Несмотря на то, что отрезок, кодируемый данным вариантом, расположен не в фолат-связывающем сайте рецептора (рисунок 4), анализ с помощью различных программ показал, что это генетическое изменение должно приводить к нестабильности белка (таблица 3). Последствие вариации – замена цистеина на тирозин в 66-й позиции – приводит к нарушению дисульфидной связи между Cys66 и Cys109, что, вероятно, изменяет трехмерную структуру рецептора FRα.
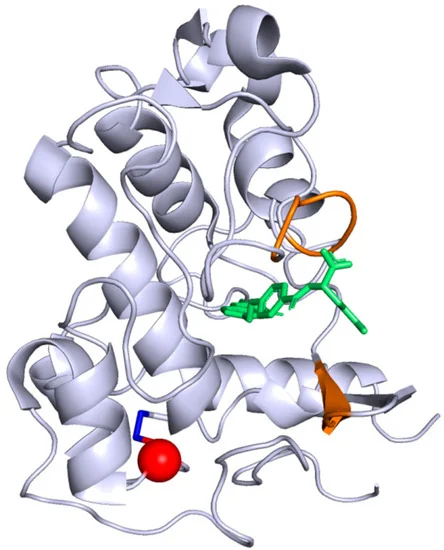
Рисунок 4. Кристаллографически определенная структура белка FRα, взятая из базы данных Protein Data Bank (4LRH). Фолат отмечен зеленым, сайт связывания фолата - оранжевым. Замена Cys66Tyr, вызванная выявленной нами патогенной вариацией, помечена красным цветом, а дисульфидная связь между Cys66 и Cys109 - темно-синим.
Таблица 2. Степень патогенности по результатам анализа в нескольких компьютерных программах, с указанием относительного балла патогенности для патогенного варианта c.197 G > A гена FOLR1.
|
Программа |
Версия |
Оценка |
Прогноз |
|
Sift [13] |
Из dbNSFP3.5 |
0.02 |
Вредная мутация |
|
Polyphen2 hvar [14] |
Из dbNSFP3.5 |
0.999 |
Предположительно повреждающая |
|
CADD phred [15] |
1.4 |
27.5 |
Входит в 0.0018% наиболее патогенных из предсказанных вариаций |
|
MutPred2 [16] |
2.0 |
0.952 |
Повреждающая мутация |
|
MutationTaster [17] |
Из dbNSFP3.5 |
1 |
Патогенная мутация |
Таблица 3. Прогноз нестабильности белка с патогенной вариацией Cys66Tyr с использованием нескольких компьютерных программ
|
Программа |
Версия |
Прогноз |
|
MUpro [18] |
1.1 |
Сниженная стабильность |
|
CUPSAT [19] |
Версия 2018.1 |
Дестабилизация |
|
AUTO-MUTE [20] |
2.0 |
Сниженная стабильность |
|
Site Directed Mutator (SDM) [21] |
Сессия связи с сервером: 02/21/2020 |
Сниженная стабильность |
|
I-Mutant2.0 [22] |
2.0 |
Снижение стабильности |
|
mCSM [23] |
Сессия связи с сервером: 02/21/2020 |
Дестабилизация |
-
Обсуждение
Нами описан новый патогенный вариант гена FOLR1 (c.197 G > A, p.Cys66Tyr (NM_016724.2)), ассоциированный с тяжелой формой церебральной фолатной недостаточности. На сегодняшний день список известных вариаций гена FOLR1 включает в себя мутации сайтов сплайсинга, нонсенс-мутации (встречаются наиболее часто) и дупликации. Эти вариации снижают экспрессию белка FRα и приводят к потере его мембранной локализации [24]. Группа Grapp et al. [1] выдвинула предположение, что миссенсные мутации могут приводить к нестабильной конформации белка FRα вследствие потери структурно важных дисульфидных связей либо сбоев посттрансляционного процессинга. Сплайс- и стоп-мутации могут приводить, соответственно, к укорочению белка FRα и к появлению кодона терминации, преждевременно останавливающего трансляцию и вызывающего полную потерю экспрессии FRα. Дупликации могут приводить к неправильному сворачиванию белка с последующей преждевременной деградацией FRα [25].
Кристаллографически определенная структура человеческого белка FRα, использованная в нашем исследовании (PDB 4LRH) [26] содержит 6 экзонов и 218 аминокислотных остатков, образующих 6 α-спиралей и 4 β-цепей. Белок содержит восемь дисульфидных связей, это необычно много. Дисульфидные связи образуются между двумя окисленными остатками цистеина и значительно стабилизируют структуру белка. Поддержание нормальной, стабильной структуры белка FRα играет принципиальную роль в сохранении сайта связывания фолата в рабочей конфигурации, и, как следствие, в сохранении нормальной транспортной функции рецептора. В нашем случае мутация, заменяющая Cys на Tyr в позиции 66, приводит к отсутствию дисульфидной связи, что, в свою очередь, может значительно сказаться на пространственной конфигурации белка FRα. Это может привести к увеличению расстояния между двумя доменами (Cys66 расположен на второй β-цепи (позиции 63-67), а Cys109 – на четвертой α-цепи (позиции 94-109)(рисунок 4)). Вместе с тем воздействие на функцию рецептора FRα не является очевидным, поскольку дисульфидный мостик Cys66-Cys109 расположен далеко от функционального сайта рецептора. Мы предполагаем, что мутация Cys66Tyr в основном сказывается на динамике белка, что, в свою очередь, изменяет его работу. Таким образом, изменение структуры белка нарушает перенос фолата в мозг и приводит к истощению запасов 5-MTHF в мозге [27,28].
Согласно Steinfeld R. et al. [25], недостаточность 5-MTHF в мозге приводит к нарушениям синтеза миелина, что можно объяснить наличием в мозге особой цепочки реакций, основанной на передаче метильных групп и связывающей метаболизм фолата и холина. Действительно, после высвобождения метильной группы SAM преобразуется в S-аденозилгомоцистеин (SAH), являющийся метаболическим прекурсором гомоцистеина. Регенерация SAM обеспечивается переносом метильной группы от метионина, перед тем синтезированного посредством метилирования гомоцистеина за счет преобразования 5-MTHF в THF. В качестве альтернативного варианта операция метилирования может быть частью цепочки окисления холина, при этом метильная группа высвобождается после преобразования бетаина в диметилглицин. Само преобразование происходит с участием фермента бетаин-гомоцистеин S-метилтрансфераза. Эта метаболическая цепочка активируется при ЦФН, в особенности при сильном снижении концентрации 5-MTHF в СМЖ, и приводит к развитию вторичной недостаточности холина из-за формирования бетаина (рисунок 1). Поскольку холин необходим для синтеза фосфатидилхолина, фосфатидилинозитола и сфингомиелина [25], недостаточность холина приводит к повреждениям белого вещества мозга и часто вызывает истощение инозитола. Активация описанной метаболической цепочки при недостаточности 5-MTHF может служить объяснением неврологической симптоматики.
ЦФН обычно развивается в возрасте от 6 месяцев до 4.5 лет, с медианным возрастом дебюта, равным 2 годам [6]. В нашем случае неврологическое развитие ребенка протекало нормально до 18 месяцев. Наличие бессимптомного интервала в начале жизни указывает на возможное существование альтернативного транспортного механизма, не включающего рецептор FRα. Существует гипотеза о том, что рецептор FRβ компенсирует потерю функционирования рецептора FRα в первые годы жизни. Действительно, экспрессия FRβ высока во внутриутробном и послеродовом периодах, при этом данный рецептор в значительной степени гомологичен FRα (70% аминокислотной последовательности) [25].
В научной литературе содержатся сообщения об эффективности фолиниевой кислоты, назначение которой приводило к быстрой реверсии неврологических симптомов и улучшению работы мозга. Известно, что эта молекула проникает в СМЖ напрямую, используя неспецифические транспортные механизмы (PCFT и RFC), после чего может непосредственно участвовать в фолатном метаболизме [10,25,29,30]. Установлено, что чем ранее назначено лечение, тем лучше ответ на терапию [6]. Более того, стоит подчеркнуть, что при ЦФН не рекомендуется назначать фолиевую кислоту, поскольку она сильно связывается с FRα и ингибирует его возможную остаточную активность, еще более затрудняя перенос 5-MTHF в мозг [1]. В нашем случае немедленно после постановки диагноза пациенту назначили кальция фолинат в дозировке 8.9 мг/кг в сутки перорально на основании гипотезы о том, что прием кальция фолината усиливает остаточный перенос 5-MTHF в СМЖ и повышает уровни 5-MTHF в СМЖ. Одновременно с данным препаратом пациентка получала вальпроат натрия и зонисамид. Положительные эффекты были замечены на втором месяце терапии, при этом не было отмечено побочных эффектов. Судя по научной литературе, такие эффекты являются последствием нормализации уровней холина и инозитола в глиальных клетках [1]. В ходе нескольких исследований [1,31,32] комбинирование перорального приема и внутривенного введения фолиниевой кислоты оказалось более эффективно, чем исключительно пероральный прием. Наша пациентка также получала внутривенные инъекции фолината кальция – сначала раз в неделю, потом раз в месяц. В первый месяц после начала терапии было отмечено значительное повышение концентрации 5-MTHF в крови – с 2 нг/мл до 39.5 нг/мл. После двух месяцев терапии наблюдалось значительное улучшение социальных взаимодействий и неврологической симптоматики, вкупе с улучшенной координацией движений и снижением частоты миоклонических судорог, при этом не было отмечено побочных эффектов. Эти результаты указывают на эффективность фолиниевой кислоты, поскольку ее прием сопровождался снижением частоты судорог более чем на 50% [33]. В итоге для оптимизации терапии пациентка получала 500 мг/неделю кальция фолината внутривенно. Было отмечено улучшение когнитивных функций, языковых навыков и способности к ходьбе – этот факт говорит о важности ранней диагностики (таблица 4).
Таблица 4. Клиническое улучшение при терапии фолиниевой кислотой (по данным стандартного клинического осмотра).
|
До терапии |
Под воздействием терапии (2 месяца) |
|
|
Судороги |
++++ |
+ |
|
Миоклония |
+++ |
▪ менее актуально ▪ менее часто ▪ меньшее распространение → вплоть до отсутствия миоклонии |
|
Тремор |
+++ |
+ |
|
Когнитивные функции |
Не подлежат оценке (из-за судорог, миоклонии, утомляемости, слабых языковых навыков, нейромоторных навыков, и слабого взаимодействия) |
▪ улучшение |
|
Языковые навыки |
▪ затруднения речи ▪ не задает спонтанных вопросов ▪ кричит, смеется и стонет. |
▪ более беглая речь ▪ задает спонтанные вопросы ▪ слагает короткие предложения |
|
Координация движений |
▪ нарушение координации движений |
▪ улучшение координации движений: разнообразное использование рук (например, пациентка с большей легкостью держит предметы, такие как книга, переворачивает страницы; самостоятельно принимает пищу, держит карандаш, собирает башни из кубиков, перекладывает предметы из руки в руку, рисует на доске) |
|
Нейромоторные навыки |
▪ не ходит самостоятельно ▪ нуждается в кресле-коляске ▪ падения (более 2 раз в день) |
▪ улучшение ▪ ходит сама либо с частичной поддержкой ▪ прямостояние |
|
Взаимодействия |
▪ не взаимодействует ▪ не поддерживает зрительный контакт |
▪ взаимодействует с окружающими |
"+" – частота
-
Заключение
Описанный случай демонстрирует то, что церебральная фолатная недостаточность, вызванная у нашей пациентки заменой c.197 G > A в гене FOLR1, представляет собой хотя и редкое, но поддающееся терапии нейродегенеративное заболевание с дебютом в раннем детском возрасте. При появлении в первые годы жизни неврологических симптомов, типичных для этого заболевания, следует немедленно осуществить анализ концентрации 5-MTHF в спинномозговой жидкости – это поможет определить диагноз и снизить степень повреждений белого вещества мозга. Анализ конформации FRα указывает на значение цистеинового остатка (p.Cys66) в поддержании трехмерной формы и стабильности этого транспортного белка и эффективном переносе 5-MTHF в спинномозговую жидкость.
Вклад авторов
Концептуализация: S.M., F.S., F.F. и P.-A.F.; Методология: S.M., P.D., F.S., J.-F.B., F.F. и P.-A.F.; Программное обеспечение: A.-S.L. и P.D.; Формальный анализ: S.M., C.L.-R., P.C., A.-S.L., P.D., R.F., M.R., J.-F.B., F.F. и P.-A.F.; Расследование: S.M., C.L.-R., R.F., M.R., J.-F.B., F.F., и P.-A.F.; Ресурсы: S.M., P.-A.F.; Курирование данных: C.L.-R., P.C., A.-S.L., P.D., R.F., M.R., J.-F.B., F.F. и P.-A.F.; Авторская работа с текстом – составление первого чернового варианта статьи: S.M., P.D. и P.-A.F.; Авторская работа с текстом — рецензирование и редактирование, S.M., C.L.-R., P.C., A.–S.L., P.D., F.S., Y.B., R.F., M.R., J.-F.B., F.P., F.F. и P.-A.F.; Визуализация: S.M., P.-A.F.; Контроль: F.F. и P.-A.F.; Администрирование проекта: F.F. и P.-A.F. Все авторы прочитали и одобрили опубликованную версию рукописи.
Финансирование: Внешнего финансирования не потребовалось.
Благодарности: Мы благодарим Клэр Гурсо (Claire Goursaud), Флоренс Абле (Florence Hublet) и Лоика Леду (Loïc Ledoux) за великолепную техническую поддержку.
Конфликты интересов: Авторы сообщают об отсутствии конфликтов интересов.
Приложение A. Гены, включенные в панель исследования фолатного метаболизма и проанализированные с помощью NGS-методик
MTHFR (NM_005957.4), MTHFS (NM_006441.3), MTHFD1 (NM_005956.3), MTHFD2 (NM_006636.3), MTHFD1L (NM_015440.4), MTHFD2L (NM_001144978.2), FOLR1 (NM_016724.2), SLC46A1 (NM_080669.5), SLC19A1 (NM_194255.2), SLC19A2 (NM_006996.2), SLC19A3 (NM_025243.3), DHFR (NM_000791.3), FTCD (NM_006657.2), SHMT1 (NM_004169.4), SHMT2 (NM_005412.5).
Список литературы
Grapp, M.; Just, I.A.; Linnankivi, T.; Wolf, P.; Lücke, T.; Häusler, M.; Gärtner, J.; Steinfeld, R. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain 2012, 135, 2022–2031. [Google Scholar] [CrossRef]
Imbard, A.; Benoist, J.F.; Blom, H.J. Neural tube defects, folic acid and methylation. Int. J. Environ. Res. Public Health 2013, 10, 4352–4389. [Google Scholar] [CrossRef]
Frewin, R. Biochemical aspects of anaemia. In Clinical Biochemistry: Metabolic and Clinical Aspects: Third Edition; Churchill Livingstone, Elsevier: London, UK, 2014; pp. 515–532. ISBN 9780702054785. [Google Scholar]
Cario, H.; Smith, D.E.C.; Blom, H.; Blau, N.; Bode, H.; Holzmann, K.; Pannicke, U.; Hopfner, K.P.; Rump, E.M.; Ayric, Z.; et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am. J. Hum. Genet. 2011, 88, 226–231. [Google Scholar] [CrossRef]
Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef]
Pope, S.; Artuch, R.; Heales, S.; Rahman, S. Cerebral folate deficiency: Analytical tests and differential diagnosis. J. Inherit. Metab. Dis. 2019, 42, 655–672. [Google Scholar] [CrossRef]
Zhao, R.; Aluri, S.; Goldman, I.D. The proton-coupled folate transporter (PCFT-SLC46A1) and the syndrome of systemic and cerebral folate deficiency of infancy: Hereditary folate malabsorption. Mol. Aspects Med. 2017, 53, 57–72. [Google Scholar] [CrossRef]
Grapp, M.; Wrede, A.; Schweizer, M.; Hüwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Bückers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef]
Jimenez, A.R.; Naz, N.; Miyan, J.A. Altered folate binding protein expression and folate delivery are associated with congenital hydrocephalus in the hydrocephalic Texas rat. J. Cereb. Blood Flow Metab. 2019, 39, 2061–2073. [Google Scholar] [CrossRef]
Pérez-Dueñas, B.; Toma, C.; Ormazábal, A.; Muchart, J.; Sanmartí, F.; Bombau, G.; Serrano, M.; García-Cazorla, A.; Cormand, B.; Artuch, R. Progressive ataxia and myoclonic epilepsy in a patient with a homozygous mutation in the FOLR1 gene. J. Inherit. Metab. Dis. 2010, 33, 795–802. [Google Scholar] [CrossRef]
Gordon, N. Cerebral Folate Deficiency Syndromes. Dev. Med. Child Neurol. 2009, 51, 180–182. [Google Scholar] [CrossRef]
Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.; Sebat, J.; Iakoucheva, L.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 2017, 134981. [Google Scholar] [CrossRef]
Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Genet. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed]
Masso, M.; Vaisman, I.I. AUTO-MUTE: Web-based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng. Des. Sel. 2010, 23, 683–687. [Google Scholar] [CrossRef]
Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef]
Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. MCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
Al-Baradie, R.S.; Chaudhary, M.W. Diagnosis and management of cerebral folate deficiency A form of folinic Acid-Responsive seizures. Neurosciences 2014, 19, 312–316. [Google Scholar]
Steinfeld, R.; Grapp, M.; Kraetzner, R.; Dreha-Kulaczewski, S.; Helms, G.; Dechent, P.; Wevers, R.; Grosso, S.; Gärtner, J. Folate Receptor Alpha Defect Causes Cerebral Folate Transport Deficiency: A Treatable Neurodegenerative Disorder Associated with Disturbed Myelin Metabolism. Am. J. Hum. Genet. 2009, 85, 354–363. [Google Scholar] [CrossRef]
Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Yong, E.L.; Xu, H.E.; Melcher, K. Crystal Structure of Human Folate Receptor Alpha in Complex with Folic Acid ENTRY 4LRH. Available online: https://www.rcsb.org/structure/4LRH (accessed on 25 March 2020).
Gocheva, G.; Ivanova, N.; Iliev, S.; Petrova, J.; Madjarova, G.; Ivanova, A. Characteristics of a Folate Receptor-α Anchored into a Multilipid Bilayer Obtained from Atomistic Molecular Dynamics Simulations. J. Chem. Theory Comput. 2020, 16, 749–764. [Google Scholar] [CrossRef]
Della-Longa, S.; Arcovito, A. Structural and functional insights on folate receptor a (FRa) by homology modeling, ligand docking and molecular dynamics. J. Mol. Graph. Model. 2013, 44, 197–207. [Google Scholar] [CrossRef]
Karin, I.; Borggraefe, I.; Catarino, C.B.; Kuhm, C.; Hoertnagel, K.; Biskup, S.; Opladen, T.; Blau, N.; Heinen, F.; Klopstock, T. Folinic acid therapy in cerebral folate deficiency: Marked improvement in an adult patient. J. Neurol. 2017, 264, 578–582. [Google Scholar] [CrossRef]
Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef]
Kobayashi, Y.; Tohyama, J.; Akiyama, T.; Magara, S.; Kawashima, H.; Akasaka, N.; Nakashima, M.; Saitsu, H.; Matsumoto, N. Severe leukoencephalopathy with cortical involvement and peripheral neuropathy due to FOLR1 deficiency. Brain Dev. 2017, 39, 266–270. [Google Scholar] [CrossRef]
Delmelle, F.; Thöny, B.; Clapuyt, P.; Blau, N.; Nassogne, M.C. Neurological improvement following intravenous high-dose folinic acid for cerebral folate transporter deficiency caused by FOLR-1 mutation. Eur. J. Paediatr. Neurol. 2016, 20, 709–713. [Google Scholar] [CrossRef]
Romero, J.; Goldenholz, D.M. Statistical efficiency of patient data in randomized clinical trials of epilepsy treatments. Epilepsia 2020, 1–9. [Google Scholar] [CrossRef]
|
Комментарий издателя: Издательство MPDI придерживается нейтральности в отношении утверждений о границах юрисдикций в публикуемых картах, а также в отношении утверждений о принадлежности авторов к институтам. |
© 2020 (авторы) Лицензиат: MDPI, Базель, Швейцария. Настоящая статья является статьей с открытым доступом, распространяемой согласно положениям и условиям лицензии Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
08 ноября 2020 г.